Ehlers–Danlosov sindrom
| Ehlers–Danlosovi sindromi | |
|---|---|
.png) Osoba sa EDS-om prikazuje hiperelastičnost kože | |
| Klasifikacija i vanjski resursi | |
| ICD-10 | Q79.6 (ILDS Q82.817) |
| ICD-9 | 756.83 |
| DiseasesDB | 4131 |
| MedlinePlus | 001468 |
| eMedicine | derm/696 ped/654 |
| MeSH | D004535 |
| GeneReviews | Ehlers–Danlosov sindrom, klasični tip |
Definicija simptomi
[uredi | uredi izvor]Ehlers–Danlosovi sindromi (EDS) su grupa nasljednih poremećaja vezivnog tkiva[1] Simptomi mogu uključivati labave zglobove, bolove u zglobovima, zategnutu kožu i abnormalne formacije ožiljaka.[1] Sve se ovo može zapaziti već u ranom djetinjstvu.[2] Komplikacije mogu uključivati dislokaciju aorte i zglobova, akoliozu, hroničnu bol ili rani osteoartritis.[1][3]
Uzroci i dijagnoza
[uredi | uredi izvor]EDS astaje zbog varijacije više od 19 različitih gena.[1] Specifični utjecaj gena određuje vrstu EDS-a.[1] Neki slučajevi proizlaze iz nove varijacije koja se pojavljuje tokom ranog razvoja, dok se drugi nasljeđuju kao autosomno dominantno ili recesivno svojstvo.[1] Tipski, te promjene obično dovode do oštećenja u strukturi ili preradi proteina kolagena.[1] Dijagnoza se često temelji na simptomima, a potvrđuje genetičkim testiranjem ili biopsijom kože.[3] Međutim, kod ljudi se u početku može pogrešno dijagnosticirati hipohondrijaza, depresija ili sindrom hroničnog umora.[3]
Liječenje i prognoza
[uredi | uredi izvor]Za ove poremećaje nije poznato lijek.[4] L iječenje je suportivno u prirodi.[3] Fizikalna terapija i potpora mogu pomoći u jačanju mišića i podržavanju zglobova.[3] Dok neki oblici EDS dovode do normalnog longeviteta, oni koji utječu na krvne žile uglavnom smanjuju životni vijek.[4]
Epidemiologija i historija
[uredi | uredi izvor]EDS pogađa barem jednog od 5.000 ljudi širom svijeta.[1] Prognoze ovise o specifičnosti poremećaja.[3] Prekomjernu pokretljivost prvi je opisao Hipokrat, 400. godine p. n. e.[5] Sindromi su imenovani prema dva ljekara, Edvardu Ehlersu (Danska) i Henri-Alexandreu Danlosu (Francuska), koji ih je opisao na prijelazu 20. stoljeća.[6]
Znaci i simptomi
[uredi | uredi izvor]Ova skupina poremećaja utiče na vezivna tkiva u tijelu, a simptomi su najčešće prisutni u zglobovima, koži i krvnim žilama. Učinci mogu biti od blago labavih zglobova do opasnih po život kardiovaskularnih.[7] Zbog raznolikosti podtipova unutar porodice EDS-ova, simptomi se mogu jako razlikovati kod pojedinaca kojima je dijagnosticiran EDS.
Mišićnoskeletni
[uredi | uredi izvor]Simptomi mišićno-koštanog sistema uključuju hiperfleksibilne zglobove koji su nestabilni i skloni uganuću, dislokacijama, subluksacijama i hiperekstenzijama.[3][6] Može se rano pojaviti i uznapredovali osteoartritis,[8] hronična degenerativna bolest zglobova,[8] deformitet prstiju u obliku labudovog vrata,[9] i Boutonniereova deformacija prstiju. Moće se pojaviti i suzenje tetiva ili mišića .[10] Deformacije kičme, kao što su skolioza (zakrivljenost), kifoza (torakalna grba), sindrom privezane kičmene moždine i okcipitoatlantoaksialna hipermobilnost, također mogu biti prisutni,[11] kao i mjalgija (bol u mišićima) i artralgija (zglobobolja),[12] kojo može biti opasno onesposobljen. Trendelenburgov znak često se viđa, što znači da kada stoji na jednoj nozi, karlica pada na drugoj strani.[13] Osgood–Schlatterova bolest, a i bolna kvrga na koljenu je takođe je uobičajena.[14] U novorođenčadi hodanje može biti odgođeno (nakon navršenih 18 mjeseci života), a umjesto puzanja može doći do prevrtanja na dno.[15]
-
 Osoba sa EDS-om pokazuje hipermobilne prste, uključujući malformaciju "labudov vrat" na 2.-5. Prstu i hipermobilni (autostoperski) palac
Osoba sa EDS-om pokazuje hipermobilne prste, uključujući malformaciju "labudov vrat" na 2.-5. Prstu i hipermobilni (autostoperski) palac -
 Individual with EDS displaying hypermobile thumb
Individual with EDS displaying hypermobile thumb -
 Hiperpokretljivodt metakarpofalangnih zglobova kod osobe sa EDS-om
Hiperpokretljivodt metakarpofalangnih zglobova kod osobe sa EDS-om -
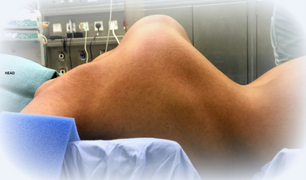 Kifoskolikoza leđa nekoga s kifoskolioznim EDS.
Kifoskolikoza leđa nekoga s kifoskolioznim EDS.




Koža
[uredi | uredi izvor]Slabo vezivno tkivo uzrokuje krhku kožu koja se lahko suzi i pomodri[8] a atrofirani ožiljci liče na papir za cigarete.[1][6][16] Pojavljuju se redundantni nabori kože, posebno na kapcima. Suvišni nabori kože područja su u kojoj višak kože leži u naborima.[8][17] Ostali simptomi kože uključuju pseudotumore mehkušce,[18] posebno na tačkama pritiska, petehijama,[19] potkožnim sferoidima,[18] livedo reticularis, a rjeđe su pijezogene papule.[20] Kod vaskularnog EDS-a koža može biti tanka i prozirna. U dermatosparaksisnom EDS-u koža je izuzetno krhka i vlažna.[1]
-
 Atrofirani ožiljak u osobe sa EDS-om
Atrofirani ožiljak u osobe sa EDS-om -
 Prozirna koža u vaskularnom EDS-u
Prozirna koža u vaskularnom EDS-u -
 Osoba sa EDS-om koji pokazuje hiperelastičnost kože
Osoba sa EDS-om koji pokazuje hiperelastičnost kože -
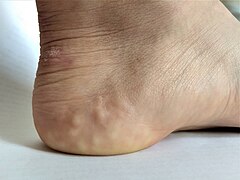 Pijezogene papule na peti osobe sa hipermobilnim EDS-om
Pijezogene papule na peti osobe sa hipermobilnim EDS-om

.png)


Kardiovaskularni
[uredi | uredi izvor]- Sindrom grudnog odvoda[21]
- Ruptura arterija
- Valvulska bolest srca, poput prolapsa mitralnog zaliska, stvara povećan rizik za infektivni endokarditis tokom operacije. To može napredovati do stupanja opasanog po život.[22] Otkrivene su nepravilnosti srčane provodljivosti kod osoba sa hipermobilnim EDS-om[23]
- Dilatacija i/ili ruptura (aneurizam) uzlazne aorte[24]
- Kardiovaskularna autonomna disfunkcija kao što je sindrom posturalne ortostatske tahikardije [25][26]
- Raynaudov fenomen
- Proširene vene[27]
- Šum na srcu
- Abnormalnosti srčane provodljivosti
Ostale manifestacije
[uredi | uredi izvor]- Hernija hiiata;
- Gastroezofagealni refluks;[28]
- Loša gastrointestinalna pokretljivost[29]
- Disautonomija[30]
- Gorlinov znak (dodirivanje vrha nosa vrhom jezika);[31]
- Analna prolapsa;[18]
- Kolaps pluća (spontani pneumotoraks);[8]
- Nervni poremećaji (sindrom karpusnog tunela, akroparestezija, neuropatija, uključujući neuropatiju malih vlakana);[32]
- Neosjetljivost na lokalne anestetike;[33]
- Arnold–Chiarijeva malformacija;[34]
- Nedostatak zgrušavanje trombocita (trombociti se ne lijepe ispravno)
- Poremećaji masnih ćelija (uključujući sindom aktivacije mastocita I mastocitozu)[35]
- Komplikacije u trudnoći: pojačan bol, blago do umjereno perparitumsko krvarenje, cervikalna insuficijencija, suzenje maternice,[10] ili preuranjena ruptura membrane.[36]
- Gluhoća; može se javiti kod nekih tipova[37]
- Oči: Kratkovidnost, kidanje mrežnjače i odvajanje mrežnjače, keratokonus, plava sklera, suho oko, Sjögrenov sindrom, subluksacija sočiva, angioidni tragovi, epicanthalni nabori , strabizam, ožiljak rožnice, sindrom lomljive rožnjače, katarakta, karotidno-kavernozne sinusne fistule, degeneracija makule;[38]
- Nestabilnost kranijskog pršljena: uzrokovana traumama (u predjelima) glave i vrata kao što su potres mozga i udarci. Ligamenti na vratu ne mogu se zaliječiti pravilno, prema tome, struktura vrata nema mogućnost održavanja lobanje, koja tada može klonuti u moždano stablo blokirajući normalan protok cerebrospinalne tečnosti, što dovodi do problema povezanih sa zatajivanjem autonomnog nervnog sistema, da radi pravilno.[39]
- Celijakija može biti povezana sa EDS. Također, može se pogrešno dijagnosticirati kao EDS zbog uobičajenih simptoma, uključujući umor, bol, gastrointestinalne tegobe ili kardiovaskularnu autonomnu disfunkciju.[22]
-
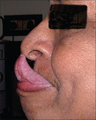 Gorlinov znak
Gorlinov znak -
 Slučaj keratoglobusa u slučaju sindroma lomljive rožnjače
Slučaj keratoglobusa u slučaju sindroma lomljive rožnjače


Zbog toga što se u djetinjstvu često dijagnosticira ili pogrešno dijagnosticira, neki slučajevi EDS-a pogrešno su okarakterizirani kao zlostavljanje djece.[40] Bol povezana sa pomenutim poremećajima može biti jaka.[41]
Genetika
[uredi | uredi izvor]
(a) Normalna kolagena vlakna su jednolične veličine i razmaka.
(b) Vlakne od osobe sa dermatosparaxisom pokazuju dramatične promjene u morfologiji fibrila s ozbiljnim efektima na vučnu čvrstoću vezivnog tkiva.
(c) Osobe s klasičnim EDS pokazuju složena vlakna.
(d) Vlakne od osobe sa nedostatkom TNX-a jednolične su veličine i ne vide se složena.
(e) TNX-null vlakna su manje pakirana i nisu dobro usklađena sa susjednim.
Svaki tip EDS-a, izuzev hipermobilnog, može biti pozitivno vezano za određene genetske varijacije. Varijacija u ovim genima može izazvati EDS:[42]
- Kolagen: primarna struktura i obrada kolagena: ADAMTS2, COL1A1, COL1A2, COL3A1, COL5A1, COL5A2
- Savijanje kolagena i poprečno povezivanje kolagena: PLOD1, FKBP14
- Struktura i funkcija miomatriksa: TNXB, COL12A1] '
- Biosinteza glikozaminoglikana: B4GALT7 , 'B3GALT6' ',' 'CHST14' DSE
- Put komplementa: C1R , C1S
- Unutarćelijski procesi: SLC39A13, ZNF469, PRDM5
Varijacije ovih gena obično mijenjaju strukturu, proizvodnju ili preradu kolagena ili proteina koji djeluju u interakciji s kolagenom. Kolagen pruža strukturu i čvrstoću vezivnom tkivu. Kvar kolagena može oslabiti vezivno tkivo u koži, kostima, krvnim žilama i organima, što rezultira karakteristikama poremećaja.[1] Obrasci nasljeđivanja ovise o specifičnom sindromu. Većina oblika EDS nasljeđuje se po autosomno dominantnom obrascu, što znači da samo jedna od dvije kopije odgovornog gena mora biti promijenjena da bi izazvala poremećaj. Nekoliko ih se nasljeđuje po autosomno recesivnom modelu, što znači da se dvije kopije gena moraju izmijeniti kako bi osoba bila pogođena poremećajem. Može biti i de novo ili sporadičnih varijacija. Sporadične varijacije nastaju bez nasljeđivanja.[43]
Dijagnoza
[uredi | uredi izvor]Dijagnoza se može postaviti evaluacijom anamneze i kliničkim posmatranjem. Beightonovi kriteriji se široko koriste za procjenu stupnja hipermobilnosti zglobova. DNK i biohemijske studije mogu pomoći u identificiranju pogođenih osoba. Dijagnostički testovi uključuju ispitivanje varijante gena za kolagen, tipizaciju kolagena kožnom biopsijom, ehokardiogramom i aktivnošću lizil hidroksilaze ili oksidaze. Međutim, ovi testovi nisu u stanju potvrditi sve slučajeve, posebno u onda kada nisu zabilježene varijacije, pa je klinička procjena i dalje važna. Ako je pogođeno više osoba u porodici, prenatalna dijagnoza može biti moguća korišćenjem DNK informacije poznate kao studija povezanosti. U toku su istraživanja radi identificiranja genetičkih markera za sve tipove.[44][45][46][47]
Poznati slučajevi/osobe
[uredi | uredi izvor]- Glumica Cherylee Houston, hipermobilni EDS.[48]
- Kraljičin garderober Yvie Oddly [49]
- Eric the Actor,[50]
- Glumica I aktivistica Jameela Jamil, hipermobilni EDS.[51] EDS is now part of her body positivity movements.[51]
- Spisateljica i glumica Lena Dunham[52]
- Australijski pjevač Sia[53]
- YouTuber i aktivista za prava invalida Annie Elainey[54]
- Učesnica na takmičenju za mis Amerike (Mis Alegany County) Victoria Graham[55]
Također pogledajte
[uredi | uredi izvor]Reference
[uredi | uredi izvor]- ^ a b c d e f g h i j k "Ehlers–Danlos syndrome". Genetics Home Reference. Arhivirano s originala, 8. 5. 2016. Pristupljeno 8. 5. 2016.
- ^ Anderson, Bryan E. (2012). The Netter Collection of Medical Illustrations - Integumentary System E-Book (2 izd.). Elsevier Health Sciences. str. 235. ISBN 978-1455726646. Arhivirano s originala, 5. 11. 2017.
- ^ a b c d e f g Lawrence EJ (decembar 2005). "The clinical presentation of Ehlers-Danlos syndrome". Advances in Neonatal Care. 5 (6): 301–14. doi:10.1016/j.adnc.2005.09.006. PMID 16338669.CS1 održavanje: upotreba parametra authors (link)
- ^ a b Ferri, Fred F. (2016). Ferri's Netter Patient Advisor. Elsevier Health Sciences. str. 939. ISBN 9780323393249. Arhivirano s originala, 5. 11. 2017. Nepoznati parametar
|name-list-format=zanemaren (prijedlog zamjene:|name-list-style=) (pomoć) - ^ Beighton, Peter H.; Grahame, Rodney; Bird, Howard A. (2011). Hypermobility of Joints. Springer. str. 1. ISBN 9781848820852. Arhivirano s originala, 5. 11. 2017. Nepoznati parametar
|name-list-format=zanemaren (prijedlog zamjene:|name-list-style=) (pomoć) - ^ a b c Byers PH, Murray ML (novembar 2012). "Heritable collagen disorders: the paradigm of the Ehlers-Danlos syndrome". The Journal of Investigative Dermatology. 132 (E1): E6-11. doi:10.1038/skinbio.2012.3. PMID 23154631.CS1 održavanje: upotreba parametra authors (link)

- ^ "Ehlers-Danlos syndrome". Genetic Home Reference. Pristupljeno 4. 4. 2018.
- ^ a b c d e "Ehlers–Danlos Syndrome". Mayo Clinic. Arhivirano s originala, 25. 6. 2012. Pristupljeno 25. 5. 2012.
- ^ Wei DH, Terrono AL (oktobar 2015). "Superficialis Sling (Flexor Digitorum Superficialis Tenodesis) for Swan Neck Reconstruction". The Journal of Hand Surgery. 40 (10): 2068–74. doi:10.1016/j.jhsa.2015.07.018. PMID 26328902.CS1 održavanje: upotreba parametra authors (link)
- ^ a b "Vascular Type-EDS". Ehlers–Danlos Syndrome Network C.A.R.E.S. Inc. Arhivirano s originala, 4. 6. 2012. Pristupljeno 25. 5. 2012.
- ^ Dordoni C, Ciaccio C, Venturini M, Calzavara-Pinton P, Ritelli M, Colombi M (august 2016). "Further delineation of FKBP14-related Ehlers-Danlos syndrome: A patient with early vascular complications and non-progressive kyphoscoliosis, and literature review" (PDF). American Journal of Medical Genetics. Part A. 170 (8): 2031–8. doi:10.1002/ajmg.a.37728. PMID 27149304.CS1 održavanje: upotreba parametra authors (link)
- ^ Gedalia A, Press J, Klein M, Buskila D (juli 1993). "Joint hypermobility and fibromyalgia in schoolchildren". Annals of the Rheumatic Diseases. 52 (7): 494–6. doi:10.1136/ard.52.7.494. PMC 1005086. PMID 8346976.CS1 održavanje: upotreba parametra authors (link)
- ^ Dommerholt, Jan (27. 1. 2012). "CSF Ehlers Danlos Colloquium, Dr Jan Dommerholt". Chiari & Syringomyelia Foundation. Arhivirano s originala, 4. 5. 2013. Pristupljeno 10. 6. 2013. Nepoznati parametar
|name-list-format=zanemaren (prijedlog zamjene:|name-list-style=) (pomoć) - ^ Vigorita, Vincent J; Mintz, Douglas; Ghelman, Bernard (2008). Orthopaedic pathology (2nd izd.). Philadelphia: Lippincott Williams and Wilkins. str. 5–6. ISBN 978-0781796705. Nepoznati parametar
|name-list-format=zanemaren (prijedlog zamjene:|name-list-style=) (pomoć) - ^ "Ehlers-Danlos syndrome - Diagnosis - Approach". BMJ Best Practice. 13. 12. 2016. Arhivirano s originala, 19. 8. 2010. Pristupljeno 18. 8. 2017.
- ^ Malfait F, Wenstrup R, De Paepe A (maj 2017). Classic Ehlers-Danlos Syndrome (Gene Reviews izd.). University of Washington.CS1 održavanje: upotreba parametra authors (link)
- ^ "Ehlers Danlos Syndrome - Morphopedics". morphopedics.wikidot.com. Pristupljeno 15. 6. 2018.
- ^ a b c "Classical Type-EDS". Ehlers–Danlos Syndrome Network C.A.R.E.S Inc. Arhivirano s originala, 30. 5. 2012. Pristupljeno 25. 5. 2012.
- ^ Portable Signs and Symptoms. Lippincott Williams & Wilkins. 2007. str. 465. ISBN 9781582556796. Arhivirano s originala, 5. 11. 2017.
- ^ "Piezogenic papules - DermNet New Zealand". www.dermnetnz.org. Arhivirano s originala, 26. 11. 2016.
- ^ Ericson WB, Wolman R (mart 2017). "Orthopaedic management of the Ehlers-Danlos syndromes". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 175 (1): 188–194. doi:10.1002/ajmg.c.31551. Provjerite vrijednost parametra
|doi=(pomoć). PMID 28192621.CS1 održavanje: upotreba parametra authors (link) - ^ a b Levy, H. P.; Adam, M. P.; Ardinger, H. H.; Pagon, R. A.; Wallace, S. E.; Bean LJH; Stephens, K.; Amemiya, A. (1993). "Hypermobile Ehlers-Danlos Syndrome". Gene Review [Internet]. University of Washington, Seattle. PMID 20301456. Nepoznati parametar
|editors=zanemaren (prijedlog zamjene:|editor=) (pomoć) - ^ Camerota F, Castori M, Celletti C, Colotto M, Amato S, Colella A, Curione M, Danese C (juli 2014). "Heart rate, conduction and ultrasound abnormalities in adults with joint hypermobility syndrome/Ehlers-Danlos syndrome, hypermobility type". Clinical Rheumatology. 33 (7): 981–7. doi:10.1007/s10067-014-2618-y. PMID 24752348. Neispravno
|display-authors=6(pomoć)CS1 održavanje: upotreba parametra authors (link) - ^ Wenstrup RJ et al. (2001). The Ehlers–Danlos Syndromes: Management of Genetic Syndromes. str. 131–149.CS1 održavanje: upotreba parametra authors (link)
- ^ Grigoriou E, Boris JR, Dormans JP (februar 2015). "Postural orthostatic tachycardia syndrome (POTS): association with Ehlers-Danlos syndrome and orthopaedic considerations". Clinical Orthopaedics and Related Research. 473 (2): 722–8. doi:10.1007/s11999-014-3898-x. PMC 4294907. PMID 25156902.CS1 održavanje: upotreba parametra authors (link)
- ^ Hakim A, O'Callaghan C, De Wandele I, Stiles L, Pocinki A, Rowe P (mart 2017). "Cardiovascular autonomic dysfunction in Ehlers-Danlos syndrome-Hypermobile type". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 175 (1): 168–174. doi:10.1002/ajmg.c.31543. PMID 28160388.CS1 održavanje: upotreba parametra authors (link)
- ^ Raffetto JD, Khalil RA (april 2008). "Mechanisms of varicose vein formation: valve dysfunction and wall dilation" (PDF). Phlebology. 23 (2): 85–98. doi:10.1258/phleb.2007.007027. PMID 18453484.CS1 održavanje: upotreba parametra authors (link)
- ^ Zeitoun JD, Lefèvre JH, de Parades V, Séjourné C, Sobhani I, Coffin B, Hamonet C (novembar 2013). "Functional digestive symptoms and quality of life in patients with Ehlers-Danlos syndromes: results of a national cohort study on 134 patients". PLOS ONE. 8 (11): e80321. Bibcode:2013PLoSO...880321Z. doi:10.1371/journal.pone.0080321. PMC 3838387. PMID 24278273.CS1 održavanje: upotreba parametra authors (link)
- ^ Brockway, Laura (april 2016). "Gastrointestinal manifestations of Ehlers–Danlos syndrome (hypermobility type)". Ehlers–Danlos Support UK. Arhivirano s originala, 14. 11. 2016. Nepoznati parametar
|name-list-format=zanemaren (prijedlog zamjene:|name-list-style=) (pomoć) - ^ "Ehlers–Danlos Syndrome". Underlying Causes of Dysautonomia. Dysautonomia International. 2012. Arhivirano s originala, 18. 12. 2014.
- ^ Létourneau Y, Pérusse R, Buithieu H (juni 2001). "Oral manifestations of Ehlers-Danlos syndrome". Journal. 67 (6): 330–4. PMID 11450296. Arhivirano s originala, 15. 12. 2016.CS1 održavanje: upotreba parametra authors (link)
- ^ "Ehlers–Danlos syndrome: Definition from". Answers.com. Arhivirano s originala, 6. 3. 2014. Pristupljeno 27. 2. 2014.
- ^ Arendt-Nielsen, Lars. "Patients Suffering from Ehlers Danlos Syndrome Type III Do Not Respond to Local Anesthetics". Arhivirano s originala, 5. 4. 2015. Nepoznati parametar
|name-list-format=zanemaren (prijedlog zamjene:|name-list-style=) (pomoć) - ^ Castori M, Voermans NC (oktobar 2014). "Neurological manifestations of Ehlers-Danlos syndrome(s): A review". Iranian Journal of Neurology. 13 (4): 190–208. PMC 4300794. PMID 25632331.CS1 održavanje: upotreba parametra authors (link)
- ^ Seneviratne SL, Maitland A, Afrin L (mart 2017). "Mast cell disorders in Ehlers-Danlos syndrome". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 175 (1): 226–236. doi:10.1002/ajmg.c.31555. PMID 28261938.CS1 održavanje: upotreba parametra authors (link)
- ^ Lind J, Wallenburg HC (april 2002). "Pregnancy and the Ehlers-Danlos syndrome: a retrospective study in a Dutch population". Acta Obstetricia et Gynecologica Scandinavica. 81 (4): 293–300. doi:10.1034/j.1600-0412.2002.810403.x. PMID 11952457.CS1 održavanje: upotreba parametra authors (link)
- ^ "Ehlers Danlos Syndromes". NORD (National Organization for Rare Disorders). Pristupljeno 11. 11. 2019.
- ^ "EHLERS-DANLOS SYNDROME – The Role of Collagen in the Eye – Information". Arhivirano s originala, 6. 7. 2019. Pristupljeno 6. 7. 2019.
- ^ Henderson, Fraser (2015). "Indices of Cranio-vertebral Instability". Funded Research. Chiari & Syringomyelia Foundation. Arhivirano s originala, 16. 9. 2016. Nepoznati parametar
|name-list-format=zanemaren (prijedlog zamjene:|name-list-style=) (pomoć) - ^ Santschi, Darrell R. (3. 4. 2008). "Redlands mother stung by untrue suspicions presses for accountability in child abuse inquiries". The Press Enterprise. Arhivirano s originala, 28. 2. 2009.
- ^ Chopra P, Tinkle B, Hamonet C, Brock I, Gompel A, Bulbena A, Francomano C (mart 2017). "Pain management in the Ehlers-Danlos syndromes". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 175 (1): 212–219. doi:10.1002/ajmg.c.31554. PMID 28186390.CS1 održavanje: upotreba parametra authors (link)
- ^ Malfait F, Francomano C, Byers P, Belmont J, Berglund B, Black J, Bloom L, Bowen JM, Brady AF, Burrows NP, Castori M, Cohen H, Colombi M, Demirdas S, De Backer J, De Paepe A, Fournel-Gigleux S, Frank M, Ghali N, Giunta C, Grahame R, Hakim A, Jeunemaitre X, Johnson D, Juul-Kristensen B, Kapferer-Seebacher I, Kazkaz H, Kosho T, Lavallee ME, Levy H, Mendoza-Londono R, Pepin M, Pope FM, Reinstein E, Robert L, Rohrbach M, Sanders L, Sobey GJ, Van Damme T, Vandersteen A, van Mourik C, Voermans N, Wheeldon N, Zschocke J, Tinkle B (mart 2017). "The 2017 international classification of the Ehlers-Danlos syndromes". American Journal of Medical Genetics. Part C, Seminars in Medical Genetics. 175 (1): 8–26. doi:10.1002/ajmg.c.31552. PMID 28306229. Neispravno
|display-authors=6(pomoć)CS1 održavanje: upotreba parametra authors (link) - ^ {{Cite news|url=https://ehlers-danlos.com/eds-types/%7Ctitle=EDS[mrtav link] Types | The Ehlers Danlos Society|work=The Ehlers Danlos Society|access-date=2017-05-22|archive-url=https://web.archive.org/web/20170624041438/https://ehlers-danlos.com/eds-types/%7Carchive-date%3D2017-06-24
- ^ Sobey G (januar 2015). "Ehlers-Danlos syndrome: how to diagnose and when to perform genetic tests". Archives of Disease in Childhood. 100 (1): 57–61. doi:10.1136/archdischild-2013-304822. PMID 24994860.CS1 održavanje: upotreba parametra authors (link)
- ^ Ross J, Grahame R (januar 2011). "Joint hypermobility syndrome". BMJ. 342: c7167. doi:10.1136/bmj.c7167. PMID 21252103.CS1 održavanje: upotreba parametra authors (link)
- ^ Castori M (2012). "Ehlers-danlos syndrome, hypermobility type: an underdiagnosed hereditary connective tissue disorder with mucocutaneous, articular, and systemic manifestations". ISRN Dermatology. 2012: 751768. doi:10.5402/2012/751768. PMC 3512326. PMID 23227356.CS1 održavanje: upotreba parametra authors (link)
- ^ "The Types of EDS". The Ehlers Danlos Society. Pristupljeno 17. 10. 2018.
- ^ "Houston hits out at "preconceived ideas" – Coronation Street News – Soaps". Digital Spy. 22. 5. 2010. Arhivirano s originala, 9. 5. 2013. Pristupljeno 27. 2. 2014.
- ^ Drag Race's Yvie Oddly On Living with Ehlers Danos Syndrome
- ^ Rosenberg, Peter; Rosenberg, Peter (30. 9. 2014). "Eric the Actor: A Eulogy". Rolling Stone. Pristupljeno 23. 7. 2019. Nepoznati parametar
|name-list-format=zanemaren (prijedlog zamjene:|name-list-style=) (pomoć) - ^ a b Gillespie, Claire. "Jameela Jamil Confirms She Has Ehlers-Danlos Syndrome". SELF (jezik: engleski). Pristupljeno 9. 8. 2019.
- ^ Lena Dunham goes on Instagram to reveal she has Ehlers-Danlos syndrome
- ^ Doherty, Jennifer (5. 10. 2019). "Ehlers-Danlos syndrome: Singer Sia's condition explained". Newsweek (jezik: engleski). Pristupljeno 11. 11. 2019.
- ^ "Here's What YouTuber Annie Elainey Wants You to Know About Being Disabled". Brit + Co (jezik: engleski). 1. 9. 2017. Pristupljeno 11. 11. 2019.
- ^ Lanquist, Lindsey. "What I Want You to Know About My Genetic Invisible Illness". SELF (jezik: engleski). Pristupljeno 11. 11. 2019.