Morbus Coats
| Klassifikation nach ICD-10 | |
|---|---|
| H35.0 | Retinopathien des Augenhintergrundes und Veränderungen der Netzhautgefäße |
| ICD-10 online (WHO-Version 2019) | |


Beim Morbus Coats (Syn. Retinitis exsudativa und retinale Teleangiektasien)[1] handelt es sich um eine seltene angeborene Augenerkrankung der Netzhautgefäße, die meistens nur einseitig auftritt und zur Verschlechterung der Sehfähigkeit, nicht selten bis zur Erblindung führt. Die Blutgefäße sind dabei erweitert und undicht, sodass Blut und lipidhaltige flüssige Absonderungen (Exsudate) in und unter die Netzhaut und ihre tieferen Schichten eindringen können. Dadurch entsteht ein Netzhautödem, welches sich von außen betrachtet durch eine weißlich-graue Pupillenfärbung (Leukokorie) bemerkbar macht. Unbehandelt kommt es infolge des Exsudats zu einer fortschreitenden entzündlichen Netzhautablösung und irreversiblen Schäden.
Bisher ist der Grund für die ursächlichen Gefäßveränderungen nicht bekannt. Der Morbus Coats tritt überwiegend bei Jungen und jungen Männern auf.[2] Die Behandlung besteht in der Verödung der Gefäße durch Kältebehandlung (Kryotherapie) oder in einer Abriegelung der retinalen Ablösungsgrenzen mittels Lasertherapie, was eine vollständige Erblindung in vielen Fällen verhindern oder zumindest deutlich hinauszögern kann. In Ausnahmefällen kann auch die Entfernung des betroffenen Auges notwendig werden.
Verbreitung
[Bearbeiten | Quelltext bearbeiten]Der Morbus Coats ist eine seltene angeborene Augenkrankheit mit sichtbaren Erweiterungen und Veränderungen (Teleangiektasien) der Netzhautgefäße. Auf 100.000 Personen kommen etwa 1–9 Erkrankungsfälle.[3] Sie treten in 90 % der Fälle einseitig auf und meist bei Knaben und jungen Männern im ersten oder zweiten Lebensjahrzehnt. Der Erkrankungsgipfel liegt zwischen sechs und acht Jahren, die Krankheit kann aber grundsätzlich alle Personen im Alter zwischen einem[4] Monat und 79 Jahren[5] befallen.[6][7][8] Dabei ist die Häufigkeit zu etwa 69 % auf männliche und zu 31 % auf weibliche Personen verteilt.[9]
Ursache und Entstehung
[Bearbeiten | Quelltext bearbeiten]Ursächlich ist ein Endotheldefekt der retinalen Blutgefäße, bei dem es zu Aussackungen und Aneurysmen dieser Gefäße kommt. Als Folge treten sowohl Untergänge der Kapillaren als auch eine Störung der Blut-Retina-Schranke auf, wodurch große Mengen Flüssigkeit (Exsudat) in die Netzhaut austreten können. Dort lagern sich auf diese Weise im Blut befindliche lipidbeladene Makrophagen und Cholesterinkristalle ab. Mit der Zeit häufen sich diese Substanzen und führen erst zur Verdickung der Netzhaut und dann letztlich zu deren – durch das Exsudat bedingten – Ablösung, mit welcher ein zunehmender Verlust der Sehkraft einhergeht.[1][6][10]
Der Grund für den ursächlichen Defekt der Netzhautgefäße ist bislang unbekannt. Bei einigen Betroffenen fanden sich erhöhte Konzentrationen eines speziellen Signalmoleküls, des Vascular Endothelial Growth Factors (VEGF).[11][12] Einzelfallbeschreibungen (Kasuistiken) über das Auftreten der Erkrankung zusammen mit unterschiedlichen anderen genetischen Defekten werden jedoch als Hinweis auf eine genetische Mitbeteiligung betrachtet.[3][13] So war in einem Fall das X-Chromosom Genort p11.4 betroffen. Dieser wird auch als NDP-Gen (siehe auch Norrie-Syndrom) bezeichnet, weil er für die Bildung von Norrin zuständig ist. Bei letzterem handelt es sich um ein Protein, von dem angenommen wird, dass es als Wachstumsfaktor Einfluss auch auf die Entwicklung der Netzhautgefäße hat.[14][15][16] Eine Assoziation mit der fazioskapulohumeralen Muskeldystrophie ist ebenfalls beschrieben. Diese Veränderung betrifft das Chromosom 4, Genort q35.[16][17][18]
Klinische Erscheinungen
[Bearbeiten | Quelltext bearbeiten]
Typische Erstsymptome sind ein sekundäres Schielen (Strabismus) und die weißlich aufleuchtende Pupille (Leukokorie). Im Falle letzterer stellt sich der Reflex des Augenhintergrunds bei mit Blitzlicht gemachten Fotografien nicht wie üblich rot, sondern weißlich-grau dar. Häufig sehen die Patienten auf dem betroffenen Auge verschwommen, wodurch auch ihr räumliches Sehen beeinträchtigt ist. Insbesondere bei Kleinkindern kann der Sehverlust auf einem Auge aber auch völlig unbemerkt bleiben. Der Morbus Coats verläuft in der Regel schmerzlos. Wenn das Exsudat allerdings zu einem Anstieg des Augeninnendruckes führt, kann daraus ein „Grüner Star“ (Glaukom) resultieren. Auch eine unterschiedliche Augenfarbe ist typisch. Der Morbus Coats verläuft in knapp zehn Prozent der Fälle anfänglich völlig asymptomatisch.[1][4][6][8]
Obgleich der Morbus Coats grundsätzlich langfristig zur Erblindung des betroffenen Auges führt, ist sein Verlauf nicht in allen Fällen gleich. Sein Fortschreiten kann spontan zu einem zeitweisen oder auch dauerhaften Stillstand kommen. Es sind einige wenige Fälle beschrieben, in denen sich die Erkrankung sogar zurückbildete. Wenn es jedoch zu einer vollständigen Netzhautablösung kommt, dann ist von einem dauerhaften Verlust der Sehkraft auszugehen. Bei Kindern unter fünf Jahren verläuft das Krankheitsbild in aller Regel wesentlich fulminanter als bei Personen über zehn Jahren.
Die überwiegende Mehrheit der Fälle entwickelt ein ausgeprägtes subretinales Exsudat und eine Ablösung der Netzhaut. Charakteristische Folgen sind dabei Gefäßneubildungen auf der Regenbogenhaut (Rubeosis iridis), Grüner und Grauer Star (Katarakt), sowie Entzündungen der mittleren Augenhaut (Uveitis) und eine Schrumpfung des Augapfels (Phthisis bulbi). Statistisch gesehen ist der untere äußere Bereich der Netzhaut besonders häufig betroffen, was zu einem medialen oberen Quadrantenausfall des Gesichtsfelds führen kann. Die Erblindungsrate ist bei denjenigen Patienten besonders hoch, bei denen sich die subretinale Flüssigkeitsansammlung nach der Behandlung nicht zurückbildet oder die Netzhaut große Zysten oder Teleangiektasien aufweist. Die Entfernung des Auges (Enukleation) wird besonders oft bei Patienten mit Grünem Star oder Rubeosis iridis notwendig. Das Krankheitsbild kann klinisch wie ein Retinoblastom imponieren.[9][19]
Liegen weitere Auffälligkeiten außerhalb des Auges im Körper vor, spricht man von einem Coats-plus-Syndrom.
Stadieneinteilungen
[Bearbeiten | Quelltext bearbeiten]Bislang wurden zwei etwas unterschiedliche Stadieneinteilungen veröffentlicht. Die erste stammt von Gomez Morales (1964) und teilt den Verlauf in fünf Stadien ein. Auf nur eng begrenzte Exsudate (Stadium I) folgen massive intraretinale (II), dann die erste kleinflächige (III) und dann die vollständige Ablösung der Netzhaut (IV). Stadium V steht für Komplikationen wie dem Glaukom.
Eine zweite Einteilung nach Shields u. a. beschreibt ebenfalls fünf zum Teil jedoch unterdifferenzierte Stadien, beginnend mit dem Stadium I, dem bloßen Vorkommen retinaler Teleangiektasien, ohne nachweislich ausgetretenem Exsudat. Stadium II beschreibt dessen Austritt außerhalb (II A) und innerhalb der Fovea centralis (II B). Stadium III beschreibt die Ablösung der Netzhaut, beginnend mit einer teilweisen Ablösung außerhalb (III A 1), bzw. innerhalb der Fovea centralis (III A 2) und der gesamten Netzhaut (III B). Stadium IV bezeichnet die vollständige Ablösung mit komplizierendem Glaukom und Stadium V ein fortgeschrittenes Endstadium der Erkrankung.[9][20]
Klinische Untersuchung
[Bearbeiten | Quelltext bearbeiten]Klassisches Leitsymptom ist die Leukokorie. Bedingt durch den einseitigen Verlust der Sehkraft und eine daraus resultierende Störung des beidäugigen Sehens (Binokularsehen) kann sich ein sekundäres Schielen entwickeln. Bei der Untersuchung des Augenhintergrundes mittels Ophthalmoskopie ist das Kapillarmuster vergröbert und die Gefäße der Netzhaut stellen sich erweitert (dilatiert) und geschlängelt dar. Dieser Befund ist meist besonders im schläfenseitigen und peripheren Bereich deutlich zu erkennen.[6] Ist die Erkrankung ausgebrochen, finden sich Netzhautablösungen, großflächige, lipidhaltig Exsudate und Blutungen aus den veränderten Gefäßen. Diese Gefäßveränderungen sind – sofern zur Diagnosefindung nötig – mittels Fluoreszenzangiographie besonders deutlich darstellbar.[1][6][21]
Technische Untersuchungsbefunde
[Bearbeiten | Quelltext bearbeiten]
Bildgebende Verfahren wie Sonographie, Computertomographie (CT) und Magnetresonanztomographie (MRT) können zur Diagnosefindung beitragen. Sonographisch imponiert der Morbus Coats als Echogenität im hinteren Bereich des Glaskörpers ohne Schallschatten; Einblutungen in den Glaskörper und die Netzhaut sind typisch.[22][23]
Im CT stellt sich der Augapfel aufgrund des eiweißhaltigen Exsudats dichter (hyperdens) im Vergleich zum Gesunden dar. Das Exsudat kann im fortgeschrittenen Stadium den gesamten Glaskörper betreffen. Der vordere Rand des subretinalen Exsudates stellt sich durch eine Kontraststeigerung dar. Da die Netzhaut um die Papille des Sehnerven herum fixiert ist, sind fortgeschrittene Ablösungen V-förmig.[6]
Im MRT weist das die Netzhaut ablösende Exsudat sowohl in den T1- als auch in den T2-gewichteten Aufnahmen eine hohe Signalintensität auf. Bei Vorliegen einer hämorrhagischen Fibrose kann es sich unregelmäßig (heterogen) darstellen. Der Raum hinter der Netzhaut erweitert sich nicht beim Einsatz von gadoliniumhaltigen Kontrastmitteln. Eine leichte Aufweitung kann sich jedoch zwischen Exsudat und dem übriggebliebenen Glaskörper zeigen. Das Exsudat weist bei der Kernspinresonanzspektroskopie eine ausgedehnte Spitze bei 1–1,6 ppm auf.[24]
Differentialdiagnose
[Bearbeiten | Quelltext bearbeiten]Differenzialdiagnostisch sind insbesondere das Retinoblastom, aber auch die Retinopathia praematurorum, ein Persistierender hyperplastischer primärer Vitreus und die Toxocariasis-bedingte Chorioretinitis, die idiopathische juxtafoveolare Teleangiektasie und die Schädigung der Netzhaut (Retinopathie) z. B. infolge Leberscher Miliaraneurysmen (unregelmäßige Ektasien der Netzhautgefäße) zu berücksichtigen,[1] die Familiäre exsudative Vitreoretinopathie, auch das Diktyom. Es sei ergänzend angemerkt, dass die Leberschen Miliaraneurysmen und der Morbus Coats sehr ähnliche Krankheitsbilder sind und daher von manchen Autoren gleichgesetzt werden.[9]
Pathologie
[Bearbeiten | Quelltext bearbeiten]- Pathologische Präparate beim Morbus Coats
-
 V-förmige Ablösung der Netzhaut durch das Exsudat.
V-förmige Ablösung der Netzhaut durch das Exsudat. -
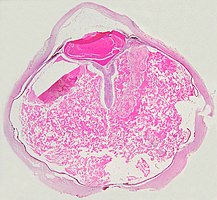 Vollständige Ablösung der Netzhaut durch das Exsudat.
Vollständige Ablösung der Netzhaut durch das Exsudat.


Eine ausgeprägte Netzhautablösung und ein gelbliches Exsudat unter der Netzhaut, das Cholesterinkristalle enthält, sind charakteristisch für den pathologischen Befund.
Unter dem Mikroskop kann die Wand der Netzhautgefäße in manchen Fällen verdickt, in anderen ausgedünnt erscheinen. Hinzu kommt eine unregelmäßige Erweiterung der betroffenen Gefäße.[25] Charakteristisch ist ein Exsudat, das sowohl aus Cholesterinkristallen, mit Cholesterin und Pigmenten beladenen Makrophagen, als auch aus roten Blutkörperchen und Hämosiderin besteht.[26] An der Netzhaut können sich eine durch das Exsudat ausgelöste granulomatöse Reaktion und in manchen Fällen auch eine durch die Verletzung ausgelöste Gliose finden.[27]
Vorbeugung, Behandlung und Heilungsaussicht
[Bearbeiten | Quelltext bearbeiten]Vorbeugende Maßnahmen sind nicht bekannt. Insbesondere im Frühstadium der Erkrankung können Kältebehandlungen (Kryotherapie) und fokale Laserkoagulation eingesetzt werden, um die veränderten Gefäße zu zerstören (veröden) und so dort absehbaren Blut- bzw. Flüssigkeitsaustritten vorzubeugen. Ist die Erkrankung bereits weiter fortgeschritten und es ist zur Netzhautablösung gekommen, können Therapieversuche in Form einer teilweisen Entfernung des Glaskörpers (Vitrektomie) oder einer Netzhautkoagulation (Verbindung mit der Aderhaut mittels Laserkoagulation) unternommen werden. Ist die Erblindung bereits eingetreten, kann auch die Entfernung (Enukleation) des betroffenen Auges notwendig sein – insbesondere dann, wenn es zu Schmerz und anderen Komplikationen kommt oder ein Retinoblastom nicht mit letzter Sicherheit ausgeschlossen werden kann. Insbesondere im Frühstadium kann oft durch den prompten Einsatz geeigneter Therapiemaßnahmen zumindest ein Teil des Sehvermögens erhalten werden. Eine kurative Therapie ist zwar nicht möglich, in einzelnen Fällen ist jedoch eine spontane Rückbildung der Erkrankung beschrieben.[1][3][8] Unter der Vorstellung, dass der Vascular Endothelial Growth Factor eine nennenswerte Bedeutung für den Verlauf der Erkrankung hat, wurde in jüngster Vergangenheit in einzelnen Fällen eine experimentelle Behandlung mittels Bevacizumab oder Pegaptanib beschrieben. Diese Vorgehensweise ist jedoch durchaus kritisch zu bewerten.[11][12][28][29][30]
Geschichte
[Bearbeiten | Quelltext bearbeiten]Die Erkrankung wurde nach ihrem Entdecker, dem schottischen Augenarzt George Coats, benannt. Er beschrieb sie im Jahre 1908 an sechs Kindern.[16][31] Im Jahre 1912 beschrieb der deutsche Augenarzt Theodor Carl Gustav von Leber[32] eine Erkrankung, die durch retinale Degeneration aufgrund von multiplen retinalen Aneurysmen charakterisiert war und vornehmlich bei jungen Männern auftrat. 1955 zeigte Reese Gemeinsamkeiten beider Krankheiten (Morbus Coats und Lebersche miliare Aneurysmen) auf und ging davon aus, dass beide lediglich verschiedene Ausprägungen derselben zugrundeliegenden Störung seien. Den Begriff Morbus Coats verwandte er dabei für die Kombination von Teleangiektasien und Retinitis exsudativa.[33]
Weblinks
[Bearbeiten | Quelltext bearbeiten]- Morbus Coats. In: Online Mendelian Inheritance in Man. (englisch)
- Katherine B Sims: NDP-Related Retinopathies. GeneReviews-Eintrag auf NCBI
- Alessandra Del Longo: Coats Disease. (PDF; 114 kB) Orphanet Encyclopedia, September 2004
Einzelnachweise
[Bearbeiten | Quelltext bearbeiten]- ↑ a b c d e f F. Grehn: Augenheilkunde. Verlag Springer, 2008, ISBN 3-540-75264-1, S. 228, eingeschränkte Vorschau in der Google-Buchsuche.
- ↑ T. Axenfeld, H. Pau: Lehrbuch und Atlas der Augenheilkunde. Unter Mitarbeit von R. Sachsenweger u. a., Gustav Fischer Verlag, Stuttgart 1980, ISBN 3-437-00255-4, S. 394.
- ↑ a b c Eintrag zu Coats-Krankheit. In: Orphanet (Datenbank für seltene Krankheiten)
- ↑ a b J. A. Shields, C. L. Shields, S. G. Honavar, H. Demirci: Clinical variations and complications of Coats disease in 150 cases. The 2000 Sanford Gifford Memorial Lecture. In: American journal of ophthalmology Band 131, Nummer 5, Mai 2001, S. 561–571, ISSN 0002-9394, PMID 11336930.
- ↑ L. M. Smithen, G. C. Brown, A. J. Brucker, L. A. Yannuzzi, C. M. Klais, R. F. Spaide: Coats’ disease diagnosed in adulthood. In: Ophthalmology. Band 112, Nummer 6, Juni 2005, S. 1072–1078, ISSN 1549-4713, doi:10.1016/j.ophtha.2004.12.038, PMID 15882905.
- ↑ a b c d e f D. P. Edward, M. F. Mafee, E. Garcia-Valenzuela, R. A. Weiss: Coats’ disease and persistent hyperplastic primary vitreous. Role of MR imaging and CT. In: Radiologic clinics of North America. Band 36, Nummer 6, November 1998, x, S. 1119–31, ISSN 0033-8389, PMID 9884692 (Review).
- ↑ A. C. Woods, J. R. Duke: Coats’s disease. I. Review of the literature, diagnostic criteria, clinical findings, and plasma lipid studies. In: The British journal of ophthalmology. Band 47, Juli 1963, S. 385–412, ISSN 0007-1161, PMID 14189710, PMC 505826 (freier Volltext).
- ↑ a b c H. Heimann u. a.: Atlas of fundus angiography. Georg Thieme Verlag, 2006, ISBN 3-13-136491-2, S. 100ff, eingeschränkte Vorschau in der Google-Buchsuche.
- ↑ a b c d A. Del Longo: Coats disease. In: Orphanet Encyclopedia, 9, 2004, orpha.net (PDF; englisch)
- ↑ M. M. Chang, I. W. McLean, J. C. Merritt: Coats’ disease. A study of 62 histologically confirmed cases. In: Journal of pediatric ophthalmology and strabismus. Band 21, Nummer 5, 1984 Sep–Oct, S. 163–168, ISSN 0191-3913, PMID 6502405.
- ↑ a b Y. G. He, H. Wang, B. Zhao, J. Lee, D. Bahl, J. McCluskey: Elevated vascular endothelial growth factor level in Coats’ disease and possible therapeutic role of bevacizumab. In: Graefe’s archive for clinical and experimental ophthalmology Band 248, Nummer 10, Oktober 2010, S. 1519–1521, ISSN 1435-702X, doi:10.1007/s00417-010-1366-1, PMID 20379736.
- ↑ a b Y. Sun, A. Jain, D. M. Moshfeghi: Elevated vascular endothelial growth factor levels in Coats disease: rapid response to pegaptanib sodium. In: Graefe’s archive for clinical and experimental ophthalmology. Band 245, Nummer 9, September 2007, S. 1387–1388, ISSN 0721-832X, doi:10.1007/s00417-007-0559-8, PMID 17342503.
- ↑ H. Heimann, u. a.: Atlas des Augenhintergrundes. Angiografie, OCT, Autofluoreszenz und Ultraschall. Georg Thieme Verlag, 2009, ISBN 3-13-146351-1, S. 142, eingeschränkte Vorschau in der Google-Buchsuche.
- ↑ OMIM Gene map: Xp11.4, NDP to Xp11.3, SLC9A7, hier online
- ↑ 0018 Norrie Disease [NDP, CYS96TRP ] dbSNP:rs104894877, hier online
- ↑ a b c Morbus Coats. In: Online Mendelian Inheritance in Man. (englisch)
- ↑ The OMIM Gene map: 4q35, FSHMD1A to 5p15.3, IRX2, hier online
- ↑ Facioscapulohumeral Muscular Dystrophy 1A; FSHMD1A, ncbi.nlm.nih.gov
- ↑ C. L. Shields, Y. Uysal, R. Benevides, R. C. Eagle, B. Malloy, J. A. Shields: Retinoblastoma in an eye with features of Coats’ disease. In: Journal of pediatric ophthalmology and strabismus. Band 43, Nummer 5, 2006 Sep–Oct, S. 313–315, ISSN 0191-3913, PMID 17022167.
- ↑ B. Förl, I. Schmack, H. E. Grossniklaus, K. Rohrschneider: Morbus Coats – Wichtige Differenzialdiagnose zum Retinoblastom. In: Der Ophthalmologe. Band 105, Nummer 8, August 2008, S. 761–764, ISSN 0941-293X, doi:10.1007/s00347-007-1645-3, PMID 18299842, PMC 299113 (freier Volltext).
- ↑ J. A. Shields, C. L. Shields: Review: coats disease. The 2001 LuEsther T. Mertz lecture. In: Retina (Philadelphia, Pa.), Band 22, Nummer 1, Februar 2002, S. 80–91, ISSN 0275-004X, PMID 11884883 (Review).
- ↑ T. Berrocal, A. de Orbe, C. Prieto, I. al-Assir, C. Izquierdo, I. Pastor, J. Abelairas: US and color Doppler imaging of ocular and orbital disease in the pediatric age group. In: Radiographics: a review publication of the Radiological Society of North America, Inc. Band 16, Nummer 2, März 1996, S. 251–272, ISSN 0271-5333, PMID 8966285.
- ↑ C. M. Glasier, M. C. Brodsky, R. E. Leithiser, S. L. Williamson, J. J. Seibert: High resolution ultrasound with Doppler. A diagnostic adjunct in orbital and ocular lesions in children. In: Pediatric radiology. Band 22, Nummer 3, 1992, S. 174–178, ISSN 0301-0449, PMID 1508582.
- ↑ L. Eisenberg, M. Castillo, L. Kwock, S. K. Mukherji, D. K. Wallace: Proton MR spectroscopy in Coats disease. In: AJNR Band 18, Nummer 4, April 1997, S. 727–729, ISSN 0195-6108, PMID 9127038.
- ↑ E. M. Chung, C. S. Specht, J. W. Schroeder: From the archives of the AFIP: Pediatric orbit tumors and tumorlike lesions: neuroepithelial lesions of the ocular globe and optic nerve. In: Radiographics Band 27, Nummer 4, 2007 Jul–Aug, S. 1159–1186, ISSN 1527-1323, doi:10.1148/rg.274075014, PMID 17620473. (Review).
- ↑ I. Kremer, I. Nissenkorn, I. Ben-Sira: Cytologic and biochemical examination of the subretinal fluid in diagnosis of Coats’ disease. In: Acta ophthalmologica. Band 67, Nummer 3, Juni 1989, S. 342–346, ISSN 0001-639X, PMID 2763826.
- ↑ B. F. Fernandes, A. N. Odashiro, S. Maloney, M. E. Zajdenweber, A. G. Lopes, M. N. Burnier: Clinical-histopathological correlation in a case of Coats’ disease. In: Diagnostic pathology. Band 1, 2006, S. 24, ISSN 1746-1596, doi:10.1186/1746-1596-1-24, PMID 16942617, PMC 1564043 (freier Volltext).
- ↑ A. Ramasubramanian, C. L. Shields: Bevacizumab for Coats’ disease with exudative retinal detachment and risk of vitreoretinal traction. In: The British journal of ophthalmology. [elektronische Veröffentlichung vor dem Druck] Juni 2011, ISSN 1468-2079, doi:10.1136/bjophthalmol-2011-300141, PMID 21653215.
- ↑ N. Goel, V. Kumar, A. Seth, U. K. Raina, B. Ghosh: Role of intravitreal bevacizumab in adult onset Coats’ disease. In: International ophthalmology. Band 31, Nummer 3, Juni 2011, S. 183–190, ISSN 1573-2630, doi:10.1007/s10792-011-9436-x, PMID 21437759.
- ↑ S. Kaul, M. Uparkar, K. Mody, J. Walinjkar, M. Kothari, S. Natarajan: Intravitreal anti-vascular endothelial growth factor agents as an adjunct in the management of Coats’ disease in children. In: Indian journal of ophthalmology. Band 58, Nummer 1, 2010 Jan–Feb, S. 76–78, ISSN 1998-3689, doi:10.4103/0301-4738.58480, PMID 20029154, PMC 284138 (freier Volltext).
- ↑ G. Coats: Forms of retinal disease with massive exudation. In: Royal London Ophthalmic Hospital Reports. Band 17, Nummer 3, 1908, S. 440–525. – Zitiert nach: Coats’ disease. whonamedit.com; abgerufen am 28. Mai 2011
- ↑ T. Leber: Über eine durch Vorkommen multipler Miliaraneurysmen charakterisierte Form der retinalen Degeneration. In: Arch Klin Ophthalmol. Band 81, 1912, S. 1–14.
- ↑ A. B. Reese: Telangiectasis of the retina and Coats’ disease. In: American journal of ophthalmology. Band 42, Nummer 1, Juli 1956, S. 1–8, ISSN 0002-9394, PMID 13339898. Zitiert nach A. Del Longo: Coats disease. In: Orphanet Encyclopedia, 9, 2004, orpha.net (PDF; englisch)