Atresia esofágica
| Atresia esofágica | ||
|---|---|---|
 Formas anatómicas de atresia del esófago: a) Atresia con fístula traqueoesofágica (Gross Tipo C); b) Atresia aislada sin fístulas (Gross Tipo A); c) Fístula en H sin atresia esofágica (Gross Tipo E) | ||
| Especialidad | genética médica | |
La atresia esofágica es un trastorno congénito caracterizado por una falta de continuidad en el trayecto del esófago, es decir, la porción superior del esófago termina abruptamente y no se continúa con la porción inferior del mismo. Se forma así un cul-de-sac («calle sin salida»)superior, vinculado con la boca, y otro inferior, que se comunica con el estómago. En la mayor parte de los casos se logra una conexión comunicante o fístula entre uno de los segmentos del esófago y la tráquea. A menudo los recién nacidos con atresia esofágica también nacen con otros trastornos congénitos del tubo digestivo, del corazón y otros órganos, a menudo no compatibles con la vida hasta el año 1939 .[1]
Clasificación
[editar]En el 98 por ciento de los casos el esófago termina en un fondo ciego con un muñón distal que comunica con el estómago y en el 86 por ciento le atraviesa una fístula que comunica con la tráquea. Con base en lo anterior, la atresia esofágica se clasifica en varios tipos:[2]
- Tipo I: atresia sin fístula (8% de los casos);
- Tipo II: con fístula en la parte superior o proximal (1% de los casos);
- Tipo III: con fístula en la parte inferior o distal (80% de los casos); los dos cul-de-sac suelen estar cerca el uno del otro;
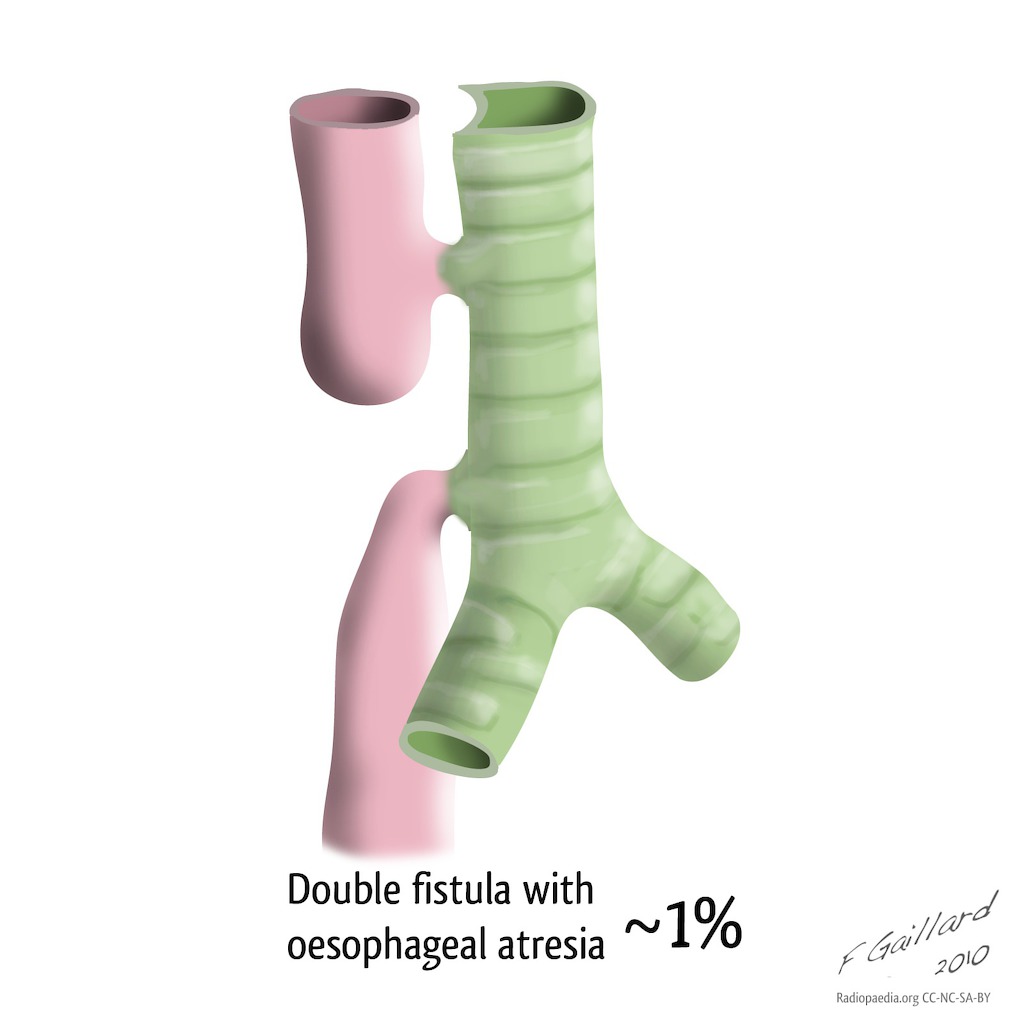
- Tipo IV: con fístula en ambas partes,
- Tipo V: con fístula en forma de H y sin atresia, en cuyo caso no se trata de una verdadera atresia, aunque se incluye como tal en la clasificación.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
El orden de la numeración varía de uno a otro autor, sin que haya variación en los tipos anatómicos de las atresias.[3]
| Gross[4] | Vogt[5] | Ladd[6] | Nombre(s) | Descripción | Frecuencia |
|---|---|---|---|---|---|
| - | Tipo 1 | - | Agenesia Esofágica | Muy rara, ausencia completa del esófago, no incluida en la clasificación de Gross o Ladd. | N/A |
| Tipo A | Tipo 2 | I | "Long Gap (Brecha amplia)", Atresia Esofágica “Pura” o “aislada" | Caracterizado por la presencia de una gran “brecha” entre las dos bolsas esofágicas ciegas sin presencia de fístula. | 7% |
| Tipo B | Tipo 3A | II | Atresia Esofágica con fístula Traqueo-Esofágica proximal | La bolsa esofágica superior se conecta de manera anormal a la tráquea. La bolsa esofágica inferior termina ciegamente. | 2-3% |
| Tipo C | Tipo 3B | III, IV | Atresia Esofágica con fístula Traqueo-Esofágica distal | La bolsa esofágica inferior se conecta de manera anormal a la tráquea. La bolsa esofágica superior termina ciegamente. | 86% |
| Tipo D | Tipo 3C | V | Atresia Esofágica con fístulas Traqueo-Esofágicas tanto proximal como distal | Tanto la bolsa esofágica superior como la inferior hacen una conexión anormal con la tráquea en dos lugares separados y aislados. | <1% |
| Tipo E | Tipo 4 | - | Fístula Traqueo-Esofágica SOLAMENTE sin Atresia Esofágica, tipo H | Esófago completamente intacto y capaz de realizar sus funciones normales, sin embargo, existe una conexión anormal entre el esófago y la tráquea. No incluido en la clasificación de Ladd | 4% |
Epidemiología
[editar]La atresia esofágica es una patología relativamente frecuente, ocurre en aproximadamente 1 por cada 3,000-4,500 nacidos vivos, frecuencia que se encuentra en descenso por razones aún desconocidas. Internacionalmente, la frecuencia registrada más elevada se encuentra en Finlandia con 1:2500 nacidos vivos.[7] Cerca del 30 por ciento de los neonatos con atresia del esófago portan una cardiopatía congénita.
Patogenia
[editar]El esófago y la tráquea se diferencian a partir de un pliegue del intestino anterior durante la cuarta semana embrionaria. Las alteraciones en el proceso de diferenciación del esófago provocan una separación incompleta del esófago y la tráquea permaneciendo una fístula. Trastornos más importantes hacen que no haya continuidad en la luz del esófago.[8] Con frecuencia existe una conexión entre la tráquea y uno de los sacos ciegos —bien sea el superior o proximal, o el inferior o distal— o en ambos.
VACTERL
[editar]La atresia esofágica suele cursar con varios síndromes congénitos reconocidos, que comprenden los designados con las siglas en inglés VACTERL:[9]
Adicionalmente, pueden asociarse trastornos cardíacos, en las extremidades, hipoplasia genital, retardo del crecimiento, anomalías del oído y sordera.[9]
Cuadro clínico
[editar]
Las indicaciones de una atresia esofágica se pueden apreciar in utero. Es característico que el feto degluta líquido amniótico durante la gestación. Aquellos sujetos con atresia esofágica son incapaces de deglutir durante el estadio fetal.[9] Esto conlleva la aparición de polihidramnios, la acumulación de una cantidad excesiva de líquido amniótico. En la valoración del recién nacido después del nacimiento, la incapacidad de introducir un catéter a través del esófago hacia el estómago es muy sugestiva de una atresia de esófago.[10]
En casos no diagnosticados en el nacimiento, el recién nacido suele presentar cianosis y dificultad respiratoria durante su primera lactancia o biberón, y en ocasiones patologías pulmonares como asfixia y neumonía.
Diagnóstico
[editar]El diagnóstico de una atresia de esófago ocurre casi siempre posnatal con la sospecha en caso de polihidramnios o en casos en donde la maniobra de inserción de la sonda nasogástrica u orogástrica del recién nacido en la sala de partos no supera los 10 cm desde la arcada dentaria hasta el cardias estomacal. De no hacerse esta maniobra al recién nacido, se observará más tarde que el bebé presenta hipersalivación, dato que pone en sospecha un probable esófago a fondo ciego que no permite que la saliva pase al estómago. Una radiografía de tórax y abdomen con contraste revela de inmediato la presencia del saco esofágico.
Pronóstico
[editar]La atresia esofágica era incompatible con la vida hasta el año 1939, cuando Height y Tawlev realizaron la primera intervención quirúrgica con resultados favorables. Desde entonces, el pronóstico ha cambiado, especialmente debido a progresos en la cirugía torácica y anestesiología. La tasa bruta de mortalidad ha disminuido desde un 100% (1939) hasta un 40%, veinte años después.[11] Actualmente la mortalidad varía de un 0%, en casos leves tratados sin retraso, hasta un 70% o más, en casos complicados con otras patologías congénitas y neonatos menores de 1,5 kg.[7]
Nuevos Tratamientos
[editar]Existe un método que permite reparar malformaciones congénitas del esófago por medio de imanes[12] sin el uso tradicional de cirugía[13] desarrollado por el Dr. Mario Zaritzky entre otros[14]
Mediante este procedimiento Annalise Dapo se convirtió en abril de 2015 en la primera paciente en los Estados Unidos en tener corregida su Atresia esofágica sin necesidad de cirugía.[15] Dapo nació sin un tercio de su esófago, lo cual le impedía comer o tragar saliva dado que su boca y estómago no estaban conectados. Esta condición es común al presentarse en 1 de cada 80.000 nacidos.[16]
Referencias
[editar]- ↑ por MedlinePlus (agosto de 2007). «Atresia esofágica». Enciclopedia médica en español. Consultado el 20 de febrero de 2009.
- ↑ Yew Shiong Leong (12 de diciembre de 2022). «Atresia de esófago». Radiopaedia.org. Consultado el 27 de abril de 2023.
- ↑ Netter, F. H., Divertie, M. B. y Brass, A. (1987) Colección Netter de ilustraciones médicas (en español). Pilar Latorre Murillo, trad. Publicado por Elsevier España, 1987, p. 111. ISBN 84-458-0220-8
- ↑ Gross RE (1953). The Surgery of Infancy and Childhood.. Philadelphia: WB Saunders.
- ↑ Vogt EC (November 1929). «Congenital esophageal atresia». American Journal of Roentgenology 22: 463-465.
- ↑ Ladd WE (1944). «The surgical treatment of esophageal atresia and tracheoesophageal fistulas». The New England Journal of Medicine 230 (21): 625-637. doi:10.1056/nejm194405252302101.
- ↑ a b Saxena, A. K; Blair, G. (abril de 2008). «Esophageal Atresia With or Without Tracheoesophageal Fistula». Infectious Diseases (en inglés). eMedicine.com. Consultado el 20 de febrero de 2009.
- ↑ Netter, F. H., Böttcher, T., Engelhardt, S. y Kortenhaus, M. Medicina interna (en español). Javier Sarmiento Martínez, trad. Publicado por Elsevier España, 2003, p. 754. ISBN 84-458-1163-0
- ↑ a b c Rustgi, A. K. Los requisitos en gastroenterología (en español). Publicado por Elsevier España, 2005, p. 3. ISBN 84-8174-821-8
- ↑ Moore, K. L. y Persaud, T. V. N. Embriología clínica (en español). Concepción Martínez Álvarez, trad. Publicado por Elsevier España, 2004, p. 257. ISBN 84-8174-725-4
- ↑ García, G., Escalada, E. y Pacheco, J. (1958). Atresia esofágica y fístulas traqueoesofágicas. Rev. Chil. Pediatr. [online], 29(7) [citado 2009-02-20], 297-306. Disponible en: < http://www.scielo.cl/scielo.php?script=sci_arttext&pid=S0370-41061958000700003&lng=es&nrm=iso >. ISSN 0370-4106.
- ↑ http://web.mit.edu/2.75/projects/DMD2011-5231.pdf
- ↑ http://www.sciencedirect.com/science/article/pii/S2213576614000372?np=y
- ↑ «Copia archivada». Archivado desde el original el 2 de abril de 2015. Consultado el 13 de abril de 2015.
- ↑ http://abc11.com/health/new-non-invasive-procedure-for-infant-at-wakemed-is-first-of-its-kind-in-us/648350/
- ↑ http://www.wral.com/pioneering-wakemed-procedure-corrects-infant-s-rare-disorder/14573393/#/vid14573546
Enlaces externos
[editar]- Asociación Española de Personas y Familiares con Atresia de Esófago http://www.atesofago.org
| Control de autoridades |
|
|---|
 Datos: Q298233
Datos: Q298233 Multimedia: Oesophageal atresia / Q298233
Multimedia: Oesophageal atresia / Q298233