Exostosis múltiple hereditaria
| Exostosis múltiple hereditaria | ||
|---|---|---|
 ftografia de las piernas de un varón de 26 años | ||
| Especialidad | genética médica | |
| Sinónimos | ||
|
Exostosis múltiple hereditaria Enfermedad de Bessel-Hagen | ||
Los osteocondromas múltiples hereditarios, también conocido como exostosis múltiples hereditarias, es un trastorno caracterizado por el desarrollo de múltiples masas osteocartilaginosas benignas (exostosis) en relación con los extremos de los huesos largos de las extremidades inferiores como el fémur y la tibia y de la miembros superiores como el húmero y los huesos del antebrazo. También se conocen como osteocondromas. Los sitios adicionales de aparición incluyen huesos planos como el hueso pélvico y la escápula. La distribución y el número de estas exostosis muestran una amplia diversidad entre los individuos afectados. Las exostosis suelen presentarse durante la infancia. La gran mayoría de las personas afectadas se manifiestan clínicamente cuando llegan a la adolescencia.[1][2] Un pequeño porcentaje de las personas afectadas tiene riesgo de desarrollar una transformación maligna, a saber, sarcomas. La incidencia de exostosis múltiples hereditarias es de alrededor de 1 de cada 50.000 individuos.[3] Los osteocondromas múltiples hereditarios es el término preferido utilizado por la Organización Mundial de la Salud.
Signos
[editar]Un bulto notable en relación con una extremidad puede ser el primer síntoma de presentación. Pueden surgir múltiples deformidades, a saber, deformidades del plano coronal alrededor de las rodillas, tobillos, hombros, codos y muñecas. Por ejemplo, se encuentran genu valgum (golpe de rodillas), tobillo valgo, arqueamiento y acortamiento cubital y subluxación de la cabeza radial. La mayoría de las personas afectadas tienen osteocondromas clínicamente manifestados alrededor de la rodilla. La participación del antebrazo en HMO es considerable.[1][4] Además, puede ocurrir baja estatura y generalmente es desproporcionada. Estas manifestaciones suelen ser el resultado de la interrupción del crecimiento de la fisis, especialmente porque los osteocondromas normalmente surgen en los extremos metafisarios de los huesos largos en las proximidades de la fisis. Los osteocondromas intraarticulares de la cadera pueden inducir limitación del rango de movimiento, dolor articular y displasia acetabular.[2] Asimismo, puede producirse dolor articular en otras localizaciones y compresión neurovascular. Además, la discapacidad funcional con respecto a las actividades de la vida diaria puede ser una característica de presentación. El dolor de la deformidad espinal o el compromiso neurológico deben despertar sospechas de afectación de las vértebras.[3]
Posible conexión con el autismo
[editar]Algunos padres de niños con HME han observado problemas sociales similares al autismo en sus hijos. Para explorar esas observaciones con mayor profundidad, un estudio de 2012 del Instituto de Investigación Médica Sanford-Burnham utilizó un modelo de ratón de HME para observar la función cognitiva. Los hallazgos indicaron que los ratones mutantes respaldaban tres características autistas: deterioro social, deterioro en la vocalización ultrasónica y comportamiento repetitivo.[5]
Genética
[editar]El HME es un trastorno hereditario autosómico dominante. Esto significa que un paciente con HME tiene un 50% de posibilidades de transmitir este trastorno a sus hijos. La mayoría de las personas con HME tienen un padre que también tiene la afección; sin embargo, aproximadamente el 10% -20% de las personas con HME tienen la afección como resultado de una mutación espontánea y, por lo tanto, son la primera persona de su familia que se ve afectada.[cita requerida]
Hasta ahora, el HME se ha relacionado con mutaciones en tres genes:
- EXT1 que se asigna al cromosoma 8q24.1[6]
- EXT2 que se asigna a 11p13[7]
- EXT3, que se asigna al brazo corto del cromosoma 19 (aunque su ubicación exacta aún no se ha determinado con precisión)[8]
Las mutaciones en estos genes normalmente conducen a la síntesis de una proteína EXT truncada que no funciona normalmente. Se sabe que las proteínas EXT son enzimas importantes en la síntesis de heparán sulfato ; sin embargo, no está claro el mecanismo exacto por el cual se alteró la síntesis de heparán sulfato que podría conducir al crecimiento óseo anormal asociado con el HME. Se cree que la proliferación y diferenciación normal de condrocitos puede verse afectada, lo que lleva a un crecimiento óseo anormal.[9][10] Dado que los genes HME están involucrados en la síntesis de un glucano ( heparán sulfato), el HME puede considerarse un trastorno congénito de la glicosilación según la nueva nomenclatura CDG sugerida en 2009.[11]
Para las personas con HME que están considerando formar una familia, las pruebas genéticas previas al implante y el diagnóstico prenatal están disponibles para determinar si el feto ha heredado la enfermedad. El HME tiene una penetrancia del 96%, lo que significa que si el gen afectado se transmite efectivamente a un niño, el niño tendrá un 96% de manifestar la enfermedad y un 4% de probabilidad de tener la enfermedad pero nunca de manifestarla. La cifra de penetrancia del 96% proviene de un solo estudio.[12] Otros estudios han observado penetrancia incompleta y variable pero sin calcular la % de penetrancia, p. ej.[13] En los dos estudios mencionados anteriormente, los individuos asintomáticos que portaban el gen defectuoso eran predominantemente mujeres, lo que llevó a la especulación de que es más probable que se presente una penetrancia incompleta en las mujeres. De hecho, otros trabajos han demostrado que los niños / hombres tienden a tener peores enfermedades que las mujeres, así como que el número de exostosis en los miembros afectados de la misma familia puede variar mucho.[14] También es posible que las mujeres se vean gravemente afectadas. La gravedad de los síntomas varía entre los individuos, incluso en la misma familia.
Es más probable que los síntomas sean graves si la mutación está en el gen ext1 en lugar de ext2 o ext3 ; ext1 es también el gen más comúnmente afectado en pacientes con este trastorno.[14]
Fisiopatología
[editar]Se caracteriza por el crecimiento de tumores óseos benignos cubiertos de cartílago alrededor de áreas de crecimiento óseo activo, particularmente la metáfisis de los huesos largos. Por lo general, se encuentran cinco o seis exostosis en las extremidades superiores e inferiores. Las ubicaciones más comunes son:[15]
El HME puede provocar el acortamiento y el arqueamiento de los huesos; las personas afectadas suelen tener una estatura baja. Dependiendo de su ubicación, las exostosis pueden causar los siguientes problemas: dolor o entumecimiento por compresión nerviosa, compromiso vascular, desigualdad en la longitud de las extremidades, irritación de tendones y músculos, deformidad de Madelung,[16] así como un rango de movimiento limitado en las articulaciones sobre que invaden. Una persona con HME tiene un mayor riesgo de desarrollar una forma poco común de cáncer de huesos llamado condrosarcoma en la edad adulta. Es posible que se presenten problemas en la vida posterior y estos podrían incluir huesos débiles y daño a los nervios.[17][18][19] La tasa de transformación informada varía desde un 0,57% [12] hasta un 8,3% de las personas con HME.[20]
Diagnóstico
[editar]El diagnóstico de HMO se basa en establecer una correlación precisa entre las características clínicas mencionadas anteriormente y las características radiográficas características. Los antecedentes familiares pueden proporcionar una pista importante para el diagnóstico. Esto se complementa con pruebas de los dos genes en los que se sabe que las variantes patogénicas causan HMO, a saber, EXT1 y EXT2. Una combinación de análisis de secuencia y análisis de deleción de todas las regiones codificantes de EXT1 y EXT2 detecta variantes patogénicas en 70 a 95% de los individuos afectados.[3][4] El sello distintivo del diagnóstico radiográfico es la presencia de osteocondromas en los extremos metafisarios de los huesos largos en los que la corteza y la médula del osteocondroma representan una extensión continua del hueso huésped. Esto es fácilmente demostrable en radiografías de rodillas.[1]
Tratamiento
[editar]Las indicaciones para la intervención quirúrgica en personas con HMO siguen sin estar claras y varían mucho en la literatura médica. En general, el tratamiento quirúrgico de HMO incluye uno o más de los siguientes procedimientos: escisión de ostecondroma, alargamiento óseo gradual o agudo como el alargamiento del cúbito, osteotomías correctivas, hemiepifisiodesis temporal para corregir deformidades de las articulaciones angulares como hemiepifisiodesis del radio distal y hemiodesisifisifisio tibial distal medial.[1][3] Sin embargo, hay poca evidencia que apoye la práctica ortopédica pediátrica en curso en los osteocondromas múltiples hereditarios. Las revisiones sistemáticas recientes encontraron evidencia insuficiente para probar que el tratamiento quirúrgico en curso de HMO mejora la función considerablemente o para probar que afecta la calidad de vida de los niños afectados.[2] Para mejorar la cantidad de evidencia en la literatura médica, se han presentado ciertas recomendaciones. La construcción de estudios prospectivos bien diseñados que puedan proporcionar una relación más clara entre los procedimientos quirúrgicos, las características del paciente y los resultados tiene una gran demanda. De lo contrario, seguir los diseños de los estudios actuales seguirá planteando más preguntas que respuestas. La artroplastia total de cadera se ha utilizado para remediar la HMO grave y dolorosa de la articulación de la cadera. La artroplastia total de cadera en personas con HMO es un desafío debido a la distorsión de la anatomía y las cirugías repetidas realizadas para abordar las quejas relacionadas con la exostosis.[21]
Epidemiología
[editar]Se estima que el HME ocurre en 1 de cada 50.000 personas.[15][19]
Imágenes adicionales
[editar]-
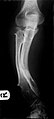 Múltiples osteocondromas que causan deformidad del antebrazo (acortamiento del radio con arqueamiento secundario del cúbito ).
Múltiples osteocondromas que causan deformidad del antebrazo (acortamiento del radio con arqueamiento secundario del cúbito ). -
 múltiples osteocondromas en la pelvis
múltiples osteocondromas en la pelvis -
 múltiples osteocondromas alrededor de la rodilla
múltiples osteocondromas alrededor de la rodilla -
 TC de osteocondroma en MO
TC de osteocondroma en MO
.jpg)
.jpg)


Referencias
[editar]- ↑ a b c d EL-Sobky, TA; Samir, S; Atiyya, AN; Mahmoud, S; Aly, AS; Soliman, R (21 de marzo de 2018). «Current paediatric orthopaedic practice in hereditary multiple osteochondromas of the forearm: a systematic review.». Sicot-J. 4: 10. PMC 5863686. PMID 29565244. doi:10.1051/sicotj/2018002.
- ↑ a b c Makhdom, AM; Jiang, F; Hamdy, RC; Benaroch, TE; Lavigne, M; Saran, N (20 de mayo de 2014). «Hip joint osteochondroma: systematic review of the literature and report of three further cases.». Advances in Orthopedics 2014: 180254. PMC 4054980. PMID 24963411. doi:10.1155/2014/180254.
- ↑ a b c d Wuyts, W (21 de noviembre de 2013). «Hereditary Multiple Osteochondromas.». GeneReviews. University of Washington, Seattle. Consultado el 24 de marzo de 2018.
- ↑ a b Alvarez, CM; De Vera, MA; Heslip, TR; Casey, B (September 2007). «Evaluation of the anatomic burden of patients with hereditary multiple exostoses.». Clin Orthop Relat Res 462: 73-79. PMID 17589361. doi:10.1097/BLO.0b013e3181334b51.
- ↑ «Autism-like socio-communicative deficits and stereotypies in mice lacking heparan sulfate». Proceedings of the National Academy of Sciences of the United States of America 109 (13): 5052-6. March 2012. Bibcode:2012PNAS..109.5052I. PMC 3323986. PMID 22411800. doi:10.1073/pnas.1117881109.
- ↑ «Genetic heterogeneity in families with hereditary multiple exostoses». American Journal of Human Genetics 53 (1): 71-9. 1993. PMC 1682231. PMID 8317501.
- ↑ «Assignment of a second locus for multiple exostoses to the pericentromeric region of chromosome 11». Human Molecular Genetics 3 (1): 167-71. 1994. PMID 8162019. doi:10.1093/hmg/3.1.167.
- ↑ «A gene for hereditary multiple exostoses maps to chromosome 19p». Human Molecular Genetics 3 (5): 717-22. 1994. PMID 8081357. doi:10.1093/hmg/3.5.717.
- ↑ «Hereditary multiple exostoses and heparan sulfate polymerization». Biochimica et Biophysica Acta (BBA) - General Subjects 1573 (3): 346-55. 2002. PMID 12417417. doi:10.1016/S0304-4165(02)00402-6.
- ↑ «Manifestations of hereditary multiple exostoses». The Journal of the American Academy of Orthopaedic Surgeons 13 (2): 110-20. 2005. PMID 15850368. doi:10.5435/00124635-200503000-00004.
- ↑ «CDG nomenclature: time for a change!». Biochim. Biophys. Acta 1792 (9): 825-6. 2009. PMC 3917312. PMID 19765534. doi:10.1016/j.bbadis.2009.08.005.
- ↑ a b «Incomplete penetrance and expressivity skewing in hereditary multiple exostoses». Clinical Genetics 52 (1): 12-6. July 1997. PMID 9272707. doi:10.1111/j.1399-0004.1997.tb02508.x.
- ↑ «Novel mutations in the EXT1 gene in two consanguineous families affected with multiple hereditary exostoses (familial osteochondromatosis)». Clinical Genetics 66 (2): 144-51. August 2004. PMID 15253765. doi:10.1111/j.1399-0004.2004.00275.x.
- ↑ a b «Severity of disease and risk of malignant change in hereditary multiple exostoses. A genotype-phenotype study». The Journal of Bone and Joint Surgery. British Volume 86 (7): 1041-6. September 2004. PMID 15446535. doi:10.1302/0301-620x.86b7.14815. Archivado desde el original el 5 de mayo de 2020. Consultado el 1 de febrero de 2021. Error en la cita: Etiqueta
<ref>no válida; el nombre «pmid15446535» está definido varias veces con contenidos diferentes - ↑ a b Turek's orthopaedics principles and their application (6th edición). Philadelphia: Lippincott Williams & Wilkins. 2005. p. 263. ISBN 9780781742986.
- ↑ Davies, A. Mark; Pettersson, Holger (2002). Pettersson, Holger; Ostensen, Harald, eds. Radiography of the Musculoskeletal System. Geneva: World Health Organization. pp. 177, 189. ISBN 978-92-4-154555-6.
- ↑ CANNON JF (1954). «Hereditary multiple exostoses». American Journal of Human Genetics 6 (4): 419-25. PMC 1716573. PMID 14349947.
- ↑ McBride WZ (September 1988). «Hereditary multiple exostoses». American Family Physician 38 (3): 191-2. PMID 3046271.
- ↑ a b «The natural history of hereditary multiple exostoses». The Journal of Bone and Joint Surgery. American Volume 76 (7): 986-92. July 1994. PMID 8027127. doi:10.2106/00004623-199407000-00005. Archivado desde el original el 20 de septiembre de 2014.
- ↑ «Chondrosarcoma in a family with multiple hereditary exostoses». The Journal of Bone and Joint Surgery. British Volume 82 (2): 261-6. March 2000. PMID 10755438. doi:10.1302/0301-620X.82B2.0820261. Archivado desde el original el 5 de mayo de 2020. Consultado el 1 de febrero de 2021.
- ↑ Vaishya, R; Swami, S; Vijay, V; Vaish, A (5 Jan 2015). «Bilateral total hip arthroplasty in a young man with hereditary multiple exostoses.». BMJ Case Rep. 2015: bcr2014207853. PMC 4289752. PMID 25564594. doi:10.1136/bcr-2014-207853.
Enlaces externos
[editar] Datos: Q1952467
Datos: Q1952467 Multimedia: Hereditary multiple exostoses / Q1952467
Multimedia: Hereditary multiple exostoses / Q1952467