BRCA2




پروتئین ۲ مستعدکننده به سرطان پستان (انگلیسی: Breast cancer type 2 susceptibility protein) که بیشتر با نام اختصاری BRCA2 شناخته میشود (بخوانید: بِـرَکا توو؛ /ˌbrækəˈtuː/[۴]) یک پروتئین است که در انسان توسط ژن «BRCA2» کُدگذاری میشود. این پروتئین با نام «بِـرَکا توو، مرتبط با ترمیم دیانای» هم شناخته میشود که این نام جدید از سوی کمیته نامگذاری ژن هوگو انتخاب شده است. پروتئین همسان آن در سایر مهرهداران «Brca2» است[۵][۶] (با حروف کوچک انگلیسی). ژن «BRCA1» در انسان، یک ژن سرکوبگر تومور است[۷][۸] و پروتئین حاصل از آن، در ترمیم آسیبهای دیانای نقش مهمی دارد.[۹]
BRCA2 و BRCA1 معمولاً در سلولهای پستان و سایر بافتها بیان میشوند و در آنجا به ترمیم دیانای آسیبدیده کمک میکنند یا اگر دیانای قابل ترمیم نباشد، سلولها را از بین میبرند. آنها در ترمیم آسیبهای کروموزومی با تأکید ویژه بر «بازسازی دیانای بدون خطا» هنگام شکستگیهای دو رشتهٔ دیانای نقش مهمی دارند.[۱۰][۱۱] اگر خود ایندو ژن حیاتی بدن در اثر جهش آسیب ببیند، دیانای آسیبدیده بهدرستی ترمیم نمیشود و این موضوع خطر ابتلا به سرطان پستان را افزایش میدهد.[۱۲][۱۳] هر دوی این ژنها به عنوان «ژنهای مستعدکننده به سرطان پستان» و «پروتئینهای مستعدکننده به سرطان پستان» توصیف شدهاند. الل غالب دارای عملکرد طبیعی سرکوب کننده تومور است در حالی که جهشهای با نفوذ بالا در این ژنها باعث از دست دادن عملکرد سرکوبگر تومور در آنها میشود که با افزایش خطر ابتلا به سرطان پستان همراه است.[۱۴]
ژن BRCA2 روی بازوی بلند کروموزوم ۱۳ در موقعیت ۱۲٫۳ قرار دارد.[۱۵] ژن مرجع BRCA2 انسانی شامل ۲۷ اگزون است و دیانای مکمل آن دارای ۱۰٬۲۵۴ جفتباز است[۱۶] که پروتئینی با ۳۴۱۸ اسید آمینه را کُدگذاری میکند.[۱۷][۱۸]
عملکرد
[ویرایش]
اگرچه ساختار ژنهای BRCA2 و BRCA1 بسیار متفاوت است، اما دستکم برخی از عملکردهای آنها به یکدیگر مرتبط هستند. پروتئینهای ساخته شده توسط هر دو ژن برای ترمیم دیانای آسیبدیده ضروری هستند (شکل مراحل ترمیم نوترکیبی را ببینید). پروتئین BRCA2 به دیانای تکرشتهای متصل میشود و بهطور مستقیم با آنزیم ریکامبیناز RAD51 برای تحریک[۲۶] و حفظ[۲۷] «یورش رشتهای»، یک مرحله حیاتی از نوترکیبی همساخت تعامل میکند. تخصیص موضعی RAD51 به شکستگیهای دورشتهای دیانای مستلزم تشکیل کمپلکس شیمیایی BRCA1-PALB2-BRCA2 است. پروتئین PALB2 (همکار و موضعیساز BRCA2)[۲۸] میتواند بهطور همافزایی با کیمارای BRCA2 (که پیکولو یا piBRCA2 هم نامیده میشود) برای گستراندن بیشتر «یورش رشتهای» همکاری کند.[۲۹] این شکستگیها ممکن است ناشی از تشعشعات طبیعی و پزشکی یا سایر عوامل آسیبرسان محیطی باشد، اما همچنین زمانی رخ میدهد که کروموزومها مواد ژنتیکی را طی یک نوع تقسیم سلولی خاص که اسپرم و تخمک ایجاد میکند (میوز) مبادله میکنند. شکستگیهای دو رشتهای نیز در طی ترمیم پیوندهای متقابل دیانای ایجاد میشود. بهطور کلی، این پروتئینها با ترمیم دیانای، در حفظ ثبات ژنوم انسان نقش دارند و از بازآرایی ژنی خطرناکی که میتواند منجر به سرطانهای خونی و سایر سرطانها شود، جلوگیری میکنند.
پژوهشگران نشان دادهاند که پروتئین BRCA2 نقش مهمی در محافظت در برابر تخریب هستهای وابسته به MRE11 توسط «چنگالهای معکوس» دارد که در طول توقف چنگال همانندسازی دیانای (ناشی از اِشکالاتی چون جهشها، مولکولهای جاگذار و غیره) تشکیل میشوند.[۳۰]
همچون BRCA1، پروتئین BRCA2 نیز احتمالاً فعالیت سایر ژنها را تنظیم میکند و نقش مهمی در رشد جنین ایفا میکند.
اهمیت بالینی
[ویرایش]
تغییرات خاصی در ژن BRCA2 خطر ابتلا به سرطان پستان را بهعنوان بخشی از سندرم ارثی سرطان پستان-تخمدان افزایش میدهد. پژوهشگران صدها جهش در این ژن را شناسایی کردهاند که بسیاری از آنها باعث افزایش خطر ابتلا به سرطان میشوند. جهشهای BRCA2 معمولاً بهصورت درج یا حذف تعداد کمی از جفتبازهای دیانای در ژن هستند. در نتیجهٔ این جهشها، محصول پروتئینی ژن BRCA2 غیرطبیعی است و به درستی عمل نمیکند. پژوهشگران بر این باورند که پروتئین معیوب BRCA2 قادر به رفع آسیب دیانای در سراسر ژنوم نیست. در نتیجه، به دلیل سنتز ترانسلیون مستعد خطا و آسیبهای دیانای ترمیمنشده، میزان جهشها افزایش مییابد و برخی از این جهشها میتوانند باعث تقسیم سلولها به روش کنترلنشده و تشکیل تومور شوند.
افرادی که دو نسخه جهش یافته از ژن BRCA2 دارند، دچار نوعی آنمی فانکونی هستند. این وضعیت به دلیل کاهش بسیار زیاد پروتئین BRCA2 در سلولها ایجاد میشود که امکان تجمع دیانای آسیب دیده را فراهم میکند. بیماران مبتلا به کمخونی فانکونی مستعد ابتلا به انواع مختلفی از سرطان خون و همچنین تومورهای توپُر بهویژه سرطانهای سر و گردن، سرطان پوست، بدخیمیهای اندامهای تناسلی؛ و نارسایی مغز استخوان (کاهش تولید سلولهای خونی که منجر به کم خونی میشود) هستند. زنانی که ژن معیوب BRCA1 یا BRCA2 را به ارث بردهاند، خطر ابتلا به سرطان پستان و سرطان تخمدان را دارند که این خطر آنچنان بالا و انتخابی است که بسیاری از حاملان جهش ژنی، جراحیهای پیشگیرانه و برداشتن تخمدان و بافت پستان را انتخاب میکنند. حدسهای زیاد و بهظاهر قابل توجهی برای توضیح چنین ویژگی بافتی وجود دارد. عوامل تعیینکننده اصلی محل وقوع سرطانهای ارثی مرتبط با BRCA1 و BRCA2 به ویژگی بافتی پاتوژن سرطان، عاملی که باعث التهاب مزمن میشود، یا عامل سرطانزا مربوط میشود. بافت هدف ممکن است گیرندههایی برای پاتوژن داشته باشد که آن را بهطور انتخابی در معرض مواد سرطانزا و یک فرایند عفونی قرار میدهد. نقص درونزاد ژنومی پاسخهای طبیعی را مختل میکند و حساسیت به بیماری را در اعضای هدف تشدید میکند. این نظریه همچنین با برخی دادهها دربارهٔ چندین سرکوبگر تومور دیگر مطابقت دارد. مزیت اصلی این مدل پشنهادی آن است که پزشکان را به فکر میاندازد که علاوه بر جراحی پیشگیرانه، گزینههای دیگری نیز وجود دارد.[۳۲]
علاوه بر سرطان پستان در مردان و زنان، جهش در ژن BRCA2 همچنین منجر به افزایش خطر ابتلا به سرطان تخمدان، لوله رحم، پروستات و لوزالمعده میشود. در برخی از مطالعات، جهش در بخش مرکزی ژن با خطر بالاتر سرطان تخمدان و خطر کمتر سرطان پروستات نسبت به جهش در سایر بخشهای ژن مرتبط بوده است. در برخی خانواده چندین نوع سرطان دیگر نیز در اثر جهش BRCA2 دیده شده است.
بهطور کلی، جهشهای شدید ارثی (از جمله جهشهای در BRCA2) تنها ۵ تا ۱۰ درصد از موارد سرطان پستان را تشکیل میدهند. میزان خطر خاص در ابتلا به سرطان پستان یا سایر سرطانها در افرادی که دچار جهش BRCA2 هستند به عوامل زیاد دیگری بستگی دارد.[۳۳]
تاریخچه
[ویرایش]ژن BRCA2 در سال ۱۹۹۴ کشف شد.[۱۵][۳۴][۳۵] در سال ۱۹۹۶، کنث آفیت و گروه پژوهشی او در مرکز سرطان مموریال اسلون–کترینگ با موفقیت شایعترین جهش را در ژن مرتبط با سرطان پستان و تخمدان در میان افراد یهودیتبار اشکنازی شناسایی کردند.[۳۶][۳۷][۳۸][۳۹]
این ژن نخستین بار توسط دانشمندان «میریاد جنتیکس»، «شرکت اِندو»، «گروه مشارکت محدود پژوهش و توسعه اچاسسی» و دانشگاه پنسیلوانیا شبیهسازی شد.[۴۰]
روشهای تشخیص احتمال ابتلا به سرطان در بیمار مبتلا به جهشهای BRCA2 و BRCA1 توسط پتنتهایی تحت مالکیت یا کنترل میریاد جنتیکس بود.[۴۱][۴۲] مدل کسبوکار میریاد جنتیکس که بهطور انحصاری آزمایش تشخیصی را ارائه میکرد، از آغاز کار آن به عنوان یک استارتآپ در سال ۱۹۹۴، منجر به تبدیل شدنش به یک شرکت سهامی عام با ۱۲۰۰ کارمند و حدود ۵۰۰ میلیون دلار درآمد سالانه در سال ۲۰۱۲ شد.[۴۳] اما در عین حال این موضوع موجب بحث و جدلهایی دربارهٔ قیمت زیاد این آزمایشها و همچنین عدم امکان تأیید نتایج توسط آزمایشگاههای تشخیصی مستقل دیگر شد که به نوبه خود منجر به شکایت «انجمن آسیبشناسی مولکولی» علیه میریاد جنتیکس شد.[۴۴]
جهشهای سلول زایا و اثر بنیانگذار
[ویرایش]تمام جهشهای BRCA2 در سلولهای زایا که تا به امروز شناسایی شدهاند، ارثی بودهاند که احتمال یک اثر بنیانگذار قابل توجه را نشان میدهد که در آن یک جهش خاص در یک گروه جمعیتی کاملاً تعریفشده مشترک است و در تئوری میتوان آن را تا یک نیای مشترک ردیابی کرد. با توجه به پیچیدگی غربالگری این تغییرات ژنی، این جهشهای مشترک ارثی ممکن است روشهای مورد نیاز برای غربالگری جهش در جمعیتهای خاص را سادهتر کنند. تجزیه و تحلیل جهشهایی که با تواتر بالا رخ میدهند نیز امکان مطالعه بیان بالینی آنها را فراهم میکند.[۴۵] نمونه بارز اثر بنیانگذار در ایسلند یافت میشود، جایی که یک جهش BRCA2 (از نوع 999del5) تقریباً در همه خانوادههای سرطان پستان/تخمدان دیده میشود.[۴۶][۴۷] این جهش دگرقالب منجر به یک محصول پروتئینی بسیار کوتاه میشود. در یک مطالعه بزرگ که صدها نفر از افراد سرطانی و افراد کنترل را مورد بررسی قرار داد، این جهش 999del5 در ۰٫۶٪ از جمعیت عمومی یافت شد. شایان ذکر است، در حالی که ۷۲ درصد از بیمارانی که اثبات شد حامل این جهش خاص هستند، سابقه خانوادگی متوسط یا قوی سرطان پستان داشتند، اما ۲۸ درصد سابقه خانوادگی کمی از سرطان داشتند یا اصلاً سابقه خانوادگی نداشتند. این یافته بهشدت نمایانگر وجود ژنهای اصلاحکنندهٔ دیگری است که بر بیان فنوتیپی این جهش یا احتمالاً برهمکنش جهش BRCA2 با عوامل محیطی تأثیر میگذارند. نمونههای دیگری از جهشهای پایهگذار در ژن BRCA2 در جدول زیر آورده شده است.
| جمعیت یا زیرگروههای قومی | جهشهای BRCA2[۴۵][۴۸] | منابع |
|---|---|---|
| یهودیان اشکنازی | 6174delT | [۴۹] |
| هلندیها | 5579insA | [۵۰] |
| فنلاندیها | 8555T>G, 999del5, IVS23-2A>G | [۵۱][۵۲] |
| کاناداییهای فرانسویتبار | 8765delAG, 3398delAAAAG | [۵۳][۵۴][۵۵] |
| مجارها | 9326insA | [۵۶] |
| ایسلندیها | 999del5 | [۴۶][۴۷] |
| ایتالیاییها | 8765delAG | [۵۷] |
| اهالی ایرلند شمالی | 6503delTT | [۵۸] |
| پاکستانیها | 3337C>T | [۵۹] |
| اسکاتلندیها | 6503delTT | [۵۸] |
| اهالی اسلوونی | IVS16-2A>G | [۶۰] |
| اسپانیاییها | 3034delAAAC(کودون 936), 9254del5 | [۶۱] |
| سوئدیها | 4486delG | [۶۲] |
میوز
[ویرایش]در گیاه آرابیدوپسیس تالیانا، از دست دادن همولوگ AtBRCA2 پروتئین BRCA2 باعث نقص شدید هم در میوز جنس نر و هم اختلال در رشد گامتوسیت ماده میشود.[۶۳] وجودِ پروتئین AtBRCA2 برای تخصیص موضعی مناسب کمپلکس سیناپتونمی پروتئین AtZYP1 و آنزیمهای ریکامبیناز AtRAD51 و AtDMC1 مورد نیاز است. علاوه بر این، AtBRCA2 برای اتصال میوزی فراخور مورد نیاز است؛ بنابراین AtBRCA2 احتمالاً برای ترمیم دیانای به شیوهٔ نوترکیبی همساخت مهم است. به نظر میرسد که AtBRCA2 برای کنترل مراحل یورش تکرشتهای با واسطه آنزیمهای AtRAD51 و AtDMC1 که جهت آسیبهای ترمیم دیانای در جریان میوز رخ میدهد، دخالت دارد.[۶۳]
همساختهای BRCA2 همچنین برای میوز در قارچ سیاهک ذرت،[۶۴] کرم الگانس[۶۵][۶۶] و مگس سرکه[۶۷] ضروری هستند.
موشهایی که نسخههای کوتاه شده و معیوب پروتئین BRCA2 را تولید میکنند، زنده اما عقیم میمانند.[۶۸] به عبارت دیگر موشهای دچار جهش BRCA2 دارای فنوتیپ توقف رشد و عقیمی در هر دو جنس هستند.[۶۹] اختلال در تولید اسپرم در این موشهای جهشیافته به دلیل شکست اتصال کروموزوم همولوگ در جریان میوز سلولی است.
توالیهای تکرارشوندهٔ بیآرسی
[ویرایش]آنزیم DMC1 (دیانای میوز ریکامبیناز ۱) یک همولوگ اختصاصی میوز از آنزیم RAD51 است که تبادل رشته در طول ترمیم به شیوهٔ نوترکیبی همساخت را میانجیگری میکند. این آنزیم در واقع تشکیل محصولات «یورش رشتهای» به دیانای (مولکولهای اتصالی) بین مولکولهای DNA همولوگ را افزایش میدهد. این آنزیم در انسان مستقیماً با هر یک از سری توالی تکرارشونده در پروتئین BRCA2 (موسم به تکرارهای بیآرسی) که تشکیل مولکولهای اتصالی توسط DMC1 را تحریک میکند، تعامل دارد.[۷۰] تکرارهای بیآرسی منطبق با یک موتیف ساختاری متشکل از یک توالی از حدود ۳۵ اسید آمینه بسیار حفاظتشده است که دستکم یک بار در همه پروتئینهای شبه BRCA2 دیده میشود. توالی بیآرسی در BRCA2 تشکیل مولکولهای اتصالی را با افزایش تعامل دیانای تکرشتهای با DMC1 تحریک میکند.[۷۰] دیانای تکرشتهای ترکیب شده شده با آنزیم DMC1 میتواند با یک دیانای تکرشتهای همولوگ از کروموزومی دیگر در مرحله خلاصه میوز جفت شود تا یک مولکول مشترک را تشکیل دهد که مرحلهای اساسی در نوترکیبی همساخت است؛ بنابراین به نظر میرسد توالیهای تکرارشوندهٔ بیآرسی در BRCA2 نقش کلیدی در ترمیم آسیبهای دیانای در جریان نوترکیبی میوزی ایفا میکنند.
عصبزایی
[ویرایش]این پروتئین در موش برای عصبزایی و جلوگیری از مدولوبلاستوما مورد نیاز است.[۷۱] از دست دادن ژن BRCA2 عمیقاً بر عصبزایی تأثیر میگذارد، به ویژه در طول رشد عصبی جنینی و پس از تولد. این نقایص عصبی از آسیب دیانای ناشی میشوند.[۷۱]
کنترل اپیژنتیک
[ویرایش]تغییرات اپیژنتیکی در بیان ژن BRCA2 (که باعث بیان بیش از حد یا کمبیانی ژن میشود) در سرطانهای غیر ارثی بسیار شایع است (جدول زیر را ببینید) در حالی که به ندرت میتوان جهش در BRCA2 را در فرد مبتلا یافت میشود.[۷۲][۷۳][۷۴]
این ژن در سرطان ریه از نوع سلول غیر کوچک، بهطور اپیژنیک توسط هیپرمتیلاسیون پروموتر سرکوب میشود.[۷۵] در این مورد، هیپرمتیلاسیون پروموتر بهطور قابل توجهی با بیان کم آرانای پیامرسان و یاخت اندک پروتئین ارتباط دارد، اما با از دست دادن هتروزیگوسیتی ژن مرتبط نیست.
در موارد غیر ارثی سرطان تخمدان اثری معکوس مشاهده میشود. پروموتر BRCA2 و نواحی رمزخوانینشدهٔ ۵ پرایم، تعداد نسبتاً کم یا حتی فاقد دینوکلئوتیدهای متیلهشدهٔ CpG در دیانای تومور در مقایسه با دیانای سلولهای سالم هستند، و ارتباط قابل توجهی بین هیپومتیلاسیون و بیان بیش از ۳ برابری ژن BRCA2 یافت میشود.[۷۶] این موضوع نشان میدهد که هیپومتیلاسیون پروموتر BRCA2 و نواحی رمزخوانینشدهٔ ۵ پرایم منجر به بیان بیش از حد آرانای پیامرسان این ژن میشود.
یک گزارش پژوهشی نشان داد که بیان ژن BRCA2 توسط ریزآرانایهای miR-146a و miR-148a کنترل اپیژنیک دارد.[۷۷]
بیان ژن BRCA2 در سرطان
[ویرایش]در یوکاریوتها، پروتئین BRCA2 نقش مهمی در ترمیم نوترکیبی همساخت دارد. در موشها و انسانها، BRCA2 اساساً همگذاری منظم RAD51 را بر روی دیانای تکرشتهای میانجیگری میکند، شکلی از مولکول که برای جفت شدن همولوگ و یورش به رشتههای دیانای فعال است.[۷۸] BRCA2 همچنین آنزیم RAD51 را از دیانای دو رشتهای تغییر مسیر میدهد و گاهی هم از جدا شدنش از دیانای دو رشتهای جلوگیری میکند.[۷۸] علاوه بر این، چهار پارالوگ آنزیم RAD51، متشکل از RAD51L1، RAD51L2، RAD51L3 و XRCC2 مجموعهای به نام «کمپلکس BCDX2» را تشکیل میدهند (شکل ترمیم نوترکیبی همساخت دیانای را ببینید). این مجموعه در بهخدمتگیری RAD51 یا تثبیت محلهای آسیب دخالت دارد.[۲۵] به نظر میرسد کمپلکس BCDX2 از طریق تسهیل همگذاری یا پایدار کردن رشته نوکلئوپروتئین RAD51 عمل میکند. آنزیم RAD51 انتقال رشته را بین یک توالی شکسته و همولوگ سالم آن تسهیل میکند تا امکان ساخت و ترمیم مجدد ناحیه آسیب دیده را فراهم کند (به مدلهای نوترکیبی همساخت مراجعه کنید).
برخی از مطالعات انجام شده بر روی سرطانها بیان بیش از حدِ ژن BRCA2 را گزارش میکنند در حالی که مطالعات دیگر بیان ناکافی آن را را گزارش کردهاند. دستکم دو گزارش بیان بیش از حد را در برخی از تومورهای غیر ارثی پستان و کمبیانی آن را در سایر تومورهای غیر ارثی پستان نشان دادند.[۷۹][۸۰] (جدول را ببینید).
بسیاری از سرطانها دارای نقص اپیژنتیکی در ژنهای مختلف ترمیم دیانای هستند (به فراوانی اپی موتاسیونها در ژنهای ترمیم دیانای در سرطانها مراجعه کنید). این نقص ترمیم احتمالاً باعث انباشت آسیبهای دیانای ترمیم نشده میشود. بیان بیش از حد BRCA2 که در بسیاری از سرطانها دیده میشود ممکن است منعکس کننده بیان جبرانی BRCA2 و افزایش ترمیم نوترکیبی همساخت باشد تا حداقل تا حدی با چنین آسیبهای اضافی دیانای مقابله کند. اِگاوا و همکاران نشان دادند که افزایش بیان BRCA2 را میتوان با بیثباتی ژنومی که اغلب در سرطانها مشاهده میشود، توضیح داد[۸۱] که بیان آرانای پیامرسان ژن BRCA2 را بهدلیل افزایش نیاز به پروتئین BRCA2 برای ترمیم دیانای تشدید میکند.
بیان ناکافی ژن BRCA2 خود منجر به انباشت آسیبهای دیانای ترمیم نشده میشود. خطاهای تکراری گذشته از این آسیبها (به سنتز ترانسلیون مراجعه کنید) منجر به افزایش جهش ژنی و بروز سرطان میشود.
| سرطان | میزان بیان ژن | میزان تغییر در بیان ژنی | روش ارزیابی | منبع. |
|---|---|---|---|---|
| سرطان تخمدان غیر ارثی | بیان بیش از حد | ۸۰٪ | آرانای پیامرسان | [۷۶] |
| سرطان تخمدان غیر ارثی | کمبیانی | ۴۲٪ | ایمونوهیستوشیمی | [۸۲] |
| (سرطانهای عودکننده/راجعه در مطالعه فوق) | افزایش بیان ژن | ۷۱٪ | ایمونوهیستوشیمی | [۸۲] |
| سرطان ریه از نوع سلول غیر کوچک | کمبیانی | ۳۴٪ | ایمونوهیستوشیمی | [۷۵] |
| سرطان پستان | بیان بیش از حد | ۶۶٪ | آرانای پیامرسان | [۸۱] |
| سرطان پستان | بیان بیش از حد | ۲۰٪ | آرانای پیامرسان | [۷۹] |
| (مشابه مطالعهٔ فوق) | کمبیانی | ۱۱٪ | آرانای پیامرسان | [۷۹] |
| سرطان پستان | بیان بیش از حد | ۳۰٪ | ایمونوهیستوشیمی | [۸۰] |
| (مشابه مطالعهٔ فوق) | کمبیانی | ۳۰٪ | ایمونوهیستوشیمی | [۸۰] |
| سرطان پستان سهگانه-منفی | کمبیانی | ۹۰٪ | ایمونوهیستوشیمی | [۸۳] |
تعاملهای شیمیایی
[ویرایش]پروتئین BRCA2 با مولکولای زیر تعامل پروتئین-پروتئین دارد:
- BRE[۸۴]
- BARD1[۸۴][۸۵]
- BCCIP[۸۶]
- BRCA1[۸۴][۸۷][۸۸][۸۹]
- BRCC3[۸۴]
- BUB1B[۹۰]
- CREBBP[۹۱]
- C11orf30[۹۲]
- FANCD2[۹۳][۹۴][۹۵]
- FANCG[۹۶]
- FLNA[۹۷]
- HMG20B[۹۸][۹۹]
- پی۵۳[۸۴][۱۰۰]
- PALB2[۲۸][۱۰۱]
- PCAF[۱۰۲][۱۰۳]
- PLK1[۱۰۲][۱۰۴]
- RAD51[۸۴][۸۷][۱۰۲][۱۰۵][۱۰۶][۱۰۷][۱۰۸][۱۰۹][۱۱۰][۱۱۱][۸۶][۸۸][۱۰۰]
- RPA1[۱۱۲]
- SHFM1[۱۱۳][۱۱۴] و
- SMAD3.[۱۱۵]
ساختار دومِـین
[ویرایش]| BRCA2 repeat | |||||||||
|---|---|---|---|---|---|---|---|---|---|
 ساختار کریستالی یک کمپلکس توالی بیآرسی rad51-brca2 | |||||||||
| شناسهها | |||||||||
| نماد | BRCA2 | ||||||||
| پیفم | PF00634 | ||||||||
| اینترپرو | IPR002093 | ||||||||
| SCOPe | 1n0w / SUPFAM | ||||||||
| |||||||||
پروتئین BRCA2 حاوی تعداد ۳۹ تکرار اسید آمینه است که برای اتصال به آنزیم RAD51 (پروتئین کلیدی در ترمیم نوترکیبی همساخت دیانای) و مقاومت در برابر درمان متیل متانسولفونات حیاتی هستند.[۱۰۰][۱۰۷][۱۰۸][۱۱۶]
دومِـین مارپیچ BRCA2 ساختاری پیچهای دارد که از یک هسته خوشهای چهار مارپیچ (آلفا ۱، آلفا ۸، آلفا ۹، آلفا ۱۰) و دو ساختار گیرهای شکل بتا (بتا ۱ تا بتا ۴) تشکیل شده است. یک بخش تقریباً ۵۰ اسید آمینهای که شامل چهار مارپیچ کوتاه (آلفا ۲ تا آلفا ۴) است، در اطراف سطح ساختار مرکزی پیچ و تاب میخورد. در پروتئین BRCA2، مارپیچهای آلفا ۹ و آلفا ۱۰ با دومِـین OB1 آن از طریق نیروی واندروالسی پیوند میخورد و شامل ریشهٔ آبگریز و آروماتیک و همچنین از طریق زنجیر جانبی و ستون اصلی و مستحکم پیوندهای هیدروژنی احاطه میشوند. این دومِـین به پروتئین ۷۰ آمینو اسیدی DSS1 (حذف شده در سندرم شکاف دست/شکاف پا) متصل میشود، که در ابتدا به عنوان یکی از سه ژنی شناسایی شد که به یک حذف ژنتیکی جایگاه کروموزومی ۱٫۵ میلیون بازی در یک سندرم ارثی ناهنجاری رشدی ارثی نسبت داده میشود.[۱۱۴]
دومِـین OB1 این ژن شکل یک چین OB را به خود میگیرد که از یک صفحه بتای پنج رشتهای بسیار خمیده تشکیل شده است که روی خود تا میخورد تا ساختاری به نام بشکه بتا ایجاد کند. OB1 دارای یک شیار کم عمق است که توسط یک وجه از صفحهٔ انحنایافته تشکیل شده است و توسط دو حلقه مرزبندی میشود، یکی بین بتا ۱ و بتا ۲ و دیگری بین بتا ۴ و بتا ۵، که امکان اتصال ضعیف دیانای تکرشتهای را فراهم میکند. این دومِـین همچنین به پروتئین ۷۰-آمینو اسیدی DSS1 (حذف شده در سندرم شکاف دست/شکاف پا) متصل میشود.[۱۱۴]
دومِـین BRCA این ژن نیز شکل یک چین OB را به خود میگیرد که از یک صفحه صفحه بتای پنج رشتهای بسیار خمیده تشکیل شده است که روی خود تا میشود تا یک بشکه بتا را تشکیل دهد. OB3 دارای یک شیار برجسته است که توسط یک وجه از صفحهٔ انحنایافته تشکیل شده است و توسط دو حلقه مرزبندی میشود، یکی بین بتا ۱ و بتا ۲ و دیگری بین بتا ۴ و بتا ۵، که امکان اتصال قوی دیانای دورشتهای را فراهم میکند.[۱۱۴]
دومِـین تاوِر (برجمانند) ساختار ثانویهای متشکل از یک جفت مارپیچ آلفای بلند و غیر موازی (ساقه) دارد که از یک کلاف سه مارپیچی (3HB) در انتهای خود حمایت میکند. 3HB حاوی یک موتیف ساختاری مارپیچ-پیچ-مارپیچ است و شبیه به دومِـینهای متصلشونده به دیانای از ریکامبیناز مکانمحور باکتریها و همچنین فاکتورهای رونویسی یوکاریوتی MYB و هومئوباکس است. دومِـین تاوِر نقش مهمی در عملکرد سرکوبگر تومور توسط این پروتئین BRCA2 دارد و برای اتصال مناسب این پروتئین به دیانای ضروری است.[۱۱۴] مطالعات نشان داد که ترکیب این دومِـین تاوِر به صورت آلوستریک توسط پروتئین کوچک "DSS1" کنترل میشود که با دومِـینهای مارپیچ، OB1 و OB2 در پروتئین BRCA2 تعامل شیمیایی دارد.[۱۱۷]
ثبت اختراع، اجرای قانون، دعوی حقوقی و اختلافنظرها
[ویرایش]یک درخواست ثبت اختراع برای ژن جداشده BRCA1 و جهشهای محرک سرطان و همچنین روشهایی برای تشخیص احتمال ابتلا به سرطان پستان، توسط دانشگاه یوتا، مؤسسه ملی علوم بهداشت محیط (NIEHS) و میریاد جنتیکس در سال ۱۹۹۴ ثبت شد.[۴۱] طی سال آتی، میریاد جنتیکس با همکاری سایر پژوهشگران، ژن BRCA2 را جداسازی و توالییابی کرد و جهشهای مربوط را شناسایی کرد و نخستین ثبت اختراع BRCA2 توسط میریاد و سایر موسسات در سال ۱۹۹۵ در ایالات متحده ثبت شد.[۴۰] میریاد دارنده مجوز انحصاری این ثبت اختراعهاست و آنها را بهشدت در ایالات متحده آمریکا در برابر آزمایشگاههای تشخیص بالینی اجرا کرده است.[۴۴] این مدل کسبوکار باعث شد تا میریاد از یک استارتآپ ساده در سال ۱۹۹۴ به یک شرکت سهامی عام با ۱۲۰۰ کارمند و حدود ۵۰۰ میلیون دلار درآمد سالانه در سال ۲۰۱۲ مبدل شود؛[۴۳] همچنین منجر به بحثهایی در مورد قیمتهای بالا آزمایشهای ژنتیکی و ناتوانی بیمار در دریافت نظر ثانویه از دیگران و تأیید آزمایشهای اولیه (از طریق انجام آزمایشهای مشابه در مکانهای دیگر) شد. آزمایشگاههای تشخیصی، که به نوبه خود منجر به شکایت انجمن آسیبشناسی مولکولی در برابر میریاد جنتیکس شد.[۴۴][۱۱۸] انقضای این ثبت اختراعها از سال ۲۰۱۴ آغاز شد.
پیتر ملدروم، مدیر اجرایی میریاد جنتیکس، اذعان کرده است که میریاد جنتیکس در اروپا «مزایای رقابتی دیگری دارد که ممکن است اجرای چنین [ثبت اختراعی] را غیرضروری کند.»[۱۱۹]
تصمیمات قانونی پیرامون ثبت اختراع زیستشناختی BRCA1 و BRCA2 بر حوزه آزمایش ژنتیک بهطور کلی تأثیر میگذارد.[۱۲۰] در ژوئن ۲۰۱۳، طی شکایت انجمن آسیبشناسی مولکولی علیه میریاد جنتیکس (شماره ۱۲–۳۹۸)، دیوان عالی ایالات متحده آمریکا به اتفاق آرا حکم داد که «یک قطعه دیانای بهطور طبیعی محصولی از طبیعت است و صرفاً به این دلیل که جدا شده است واجد شرایط ثبت اختراع نیست» و ثبت اختراع میریاد جنتیکس در مورد ژنهای BRCA1 و BRCA2 را باطل کرد. با این حال، دادگاه همچنین اعلام کرد که دستکاری یک ژن برای ایجاد چیزی که در طبیعت یافت نمیشود همچنان میتواند برای حمایت از حق ثبت اختراع واجد شرایط باشد.[۱۲۱] دادگاه فدرال استرالیا در فوریه ۲۰۱۳ به نتیجه معکوس رسید و حق ثبت اختراع میریاد جنتیکس در استرالیا برای ژن BRCA1 را در فوریه ۲۰۱۳ تأیید کرد،[۱۲۲] اما این تصمیم در حال تجدیدنظرخواهی است و درخواست تجدیدنظر شامل رسیدگی به حکم دادگاه عالی ایالات متحده خواهد بود.[۱۲۳]
منابع
[ویرایش]- ↑ ۱٫۰ ۱٫۱ ۱٫۲ GRCm38: Ensembl release 89: ENSMUSG00000041147 - Ensembl, May 2017
- ↑ "Human PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Mouse PubMed Reference:". National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ Hamel PJ (2007-05-29). "BRCA1 and BRCA2: No Longer the Only Troublesome Genes Out There". HealthCentral. Retrieved 2010-07-02.
- ↑ "OrthoMaM phylogenetic marker: BRCA2 coding sequence". Archived from the original on 2016-03-03. Retrieved 2010-02-19.
- ↑ "BRCA2 gene tree". Ensembl. May 2021
- ↑ Duncan JA, Reeves JR, Cooke TG (October 1998). "BRCA1 and BRCA2 proteins: roles in health and disease". Molecular Pathology. 51 (5): 237–47. doi:10.1136/mp.51.5.237. PMC 395646. PMID 10193517.
- ↑ Yoshida K, Miki Y (November 2004). "Role of BRCA1 and BRCA2 as regulators of DNA repair, transcription, and cell cycle in response to DNA damage". Cancer Science. 95 (11): 866–71. doi:10.1111/j.1349-7006.2004.tb02195.x. PMID 15546503. S2CID 24297965.
- ↑ Check W (2006-09-01). "BRCA: What we know now". College of American Pathologists. Retrieved 2010-08-23.
- ↑ Friedenson B (August 2007). "The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers". BMC Cancer. 7 (1): 152–162. doi:10.1186/1471-2407-7-152. PMC 1959234. PMID 17683622.
- ↑ Friedenson B (2008-06-08). "Breast cancer genes protect against some leukemias and lymphomas" (video). SciVee.
- ↑ "Breast and Ovarian Cancer Genetic Screening". Palo Alto Medical Foundation. Archived from the original on 4 October 2008. Retrieved 2008-10-11.
- ↑ Friedenson B (2007). "The BRCA1/2 pathway prevents hematologic cancers in addition to breast and ovarian cancers". BMC Cancer. 7 (1): 152. doi:10.1186/1471-2407-7-152. PMC 1959234. PMID 17683622.
- ↑ O'Donovan PJ, Livingston DM (April 2010). "BRCA1 and BRCA2: breast/ovarian cancer susceptibility gene products and participants in DNA double-strand break repair". Carcinogenesis. 31 (6): 961–7. doi:10.1093/carcin/bgq069. PMID 20400477.
- ↑ ۱۵٫۰ ۱۵٫۱ Wooster R, Neuhausen SL, Mangion J, Quirk Y, Ford D, Collins N, Nguyen K, Seal S, Tran T, Averill D (September 1994). "Localization of a breast cancer susceptibility gene, BRCA2, to chromosome 13q12-13". Science. 265 (5181): 2088–90. Bibcode:1994Sci...265.2088W. doi:10.1126/science.8091231. PMID 8091231.
- ↑ "BRCA2 breast cancer 2, early onset [Homo sapiens]". EntrezGene. National Center for Biotechnology Information, U.S. National Library of Medicine.
- ↑ "Breast cancer type 2 susceptibility protein - Homo sapiens (Human)". P51587. UniProt.
- ↑ Williams-Jones B (2002). Genetic testing for sale: Implications of commercial brca testing in Canada (Ph.D.). The University of British Columbia.
- ↑ D'Andrea AD (2010). "Susceptibility pathways in Fanconi's anemia and breast cancer". N. Engl. J. Med. 362 (20): 1909–19. doi:10.1056/NEJMra0809889. PMC 3069698. PMID 20484397.
- ↑ Sobeck A, Stone S, Landais I, de Graaf B, Hoatlin ME (2009). "The Fanconi anemia protein FANCM is controlled by FANCD2 and the ATR/ATM pathways". J. Biol. Chem. 284 (38): 25560–8. doi:10.1074/jbc.M109.007690. PMC 2757957. PMID 19633289.
- ↑ Castillo P, Bogliolo M, Surralles J (2011). "Coordinated action of the Fanconi anemia and ataxia telangiectasia pathways in response to oxidative damage". DNA Repair (Amst.). 10 (5): 518–25. doi:10.1016/j.dnarep.2011.02.007. PMID 21466974.
- ↑ Stolz A, Ertych N, Bastians H (2011). "Tumor suppressor CHK2: regulator of DNA damage response and mediator of chromosomal stability". Clin. Cancer Res. 17 (3): 401–5. doi:10.1158/1078-0432.CCR-10-1215. PMID 21088254.
- ↑ Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D'Andrea AD (2002). "S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51". Blood. 100 (7): 2414–20. doi:10.1182/blood-2002-01-0278. PMID 12239151.
- ↑ Park JY, Zhang F, Andreassen PR (2014). "PALB2: the hub of a network of tumor suppressors involved in DNA damage responses". Biochim. Biophys. Acta. 1846 (1): 263–75. doi:10.1016/j.bbcan.2014.06.003. PMC 4183126. PMID 24998779.
- ↑ ۲۵٫۰ ۲۵٫۱ Chun J, Buechelmaier ES, Powell SN (2013). "Rad51 paralog complexes BCDX2 and CX3 act at different stages in the BRCA1-BRCA2-dependent homologous recombination pathway". Mol. Cell. Biol. 33 (2): 387–95. doi:10.1128/MCB.00465-12. PMC 3554112. PMID 23149936.
- ↑ Jensen RB, Carreira A, Kowalczykowski SC (October 2010). "Purified human BRCA2 stimulates RAD51-mediated recombination". Nature. 467 (7316): 678–83. Bibcode:2010Natur.467..678J. doi:10.1038/nature09399. PMC 2952063. PMID 20729832.
- ↑ Wang CX, Jimenez-Sainz J, Jensen RB, Mazin AV (March 2019). "The Post-Synaptic Function of Brca2". Scientific Reports. 9 (1): 4554. Bibcode:2019NatSR...9.4554W. doi:10.1038/s41598-019-41054-y. PMC 6418147. PMID 30872704.
- ↑ ۲۸٫۰ ۲۸٫۱ Xia B, Sheng Q, Nakanishi K, Ohashi A, Wu J, Christ N, Liu X, Jasin M, Couch FJ, Livingston DM (June 2006). "Control of BRCA2 cellular and clinical functions by a nuclear partner, PALB2". Molecular Cell. 22 (6): 719–29. doi:10.1016/j.molcel.2006.05.022. PMID 16793542.
- ↑ Buisson R, Dion-Côté AM, Coulombe Y, Launay H, Cai H, Stasiak AZ, Stasiak A, Xia B, Masson JY (October 2010). "Cooperation of breast cancer proteins PALB2 and piccolo BRCA2 in stimulating homologous recombination". Nature Structural & Molecular Biology. 17 (10): 1247–54. doi:10.1038/nsmb.1915. PMC 4094107. PMID 20871615.
- ↑ Mijic S, Zellweger R, Chappidi N, Berti M, Jacobs K, Mutreja K, Ursich S, Ray Chaudhuri A, Nussenzweig A, Janscak P, Lopes M (October 2017). "Replication fork reversal triggers fork degradation in BRCA2-defective cells". Nature Communications (به انگلیسی). 8 (1): 859. Bibcode:2017NatCo...8..859M. doi:10.1038/s41467-017-01164-5. PMC 5643541. PMID 29038466.
- ↑ Petrucelli N, Daly MB, Pal T (December 2016) [September 1998]. "BRCA1- and BRCA2-Associated Hereditary Breast and Ovarian Cancer". In Adam MP, Ardinger HH, Pagon RA, Wallace SE, Bean LJ, Mirzaa G, Amemiya A (eds.). GeneReviews. University of Washington, Seattle. PMID 20301425.
- ↑ Levin B, Lech D, Friedenson B (2012). "Evidence that BRCA1- or BRCA2-associated cancers are not inevitable". Molecular Medicine. 18 (9): 1327–37. doi:10.2119/molmed.2012.00280. PMC 3521784. PMID 22972572.
- ↑ "High-Penetrance Breast and/or Ovarian Cancer Susceptibility Genes". National Cancer Institute. Retrieved 7 December 2012.
- ↑ Wooster R, Bignell G, Lancaster J, Swift S, Seal S, Mangion J, Collins N, Gregory S, Gumbs C, Micklem G (1995). "Identification of the breast cancer susceptibility gene BRCA2". Nature. 378 (6559): 789–792. Bibcode:1995Natur.378..789W. doi:10.1038/378789a0. PMID 8524414. S2CID 4346791.
- ↑ High-Impact Science: Tracking down the BRCA genes (Part 2) بایگانیشده در ۲۰۱۲-۰۳-۰۳ توسط Wayback Machine - Cancer Research UK science blog, 2012
- ↑ "Kenneth Offit | Breast Cancer Research Foundation | BCRF". Bcrfcure.org. 23 June 2014. Retrieved 2015-07-16.
- ↑ "A revolution at 50; kenneth offit". The New York Times. 2003-02-25. ISSN 0362-4331. Retrieved 2015-07-02.
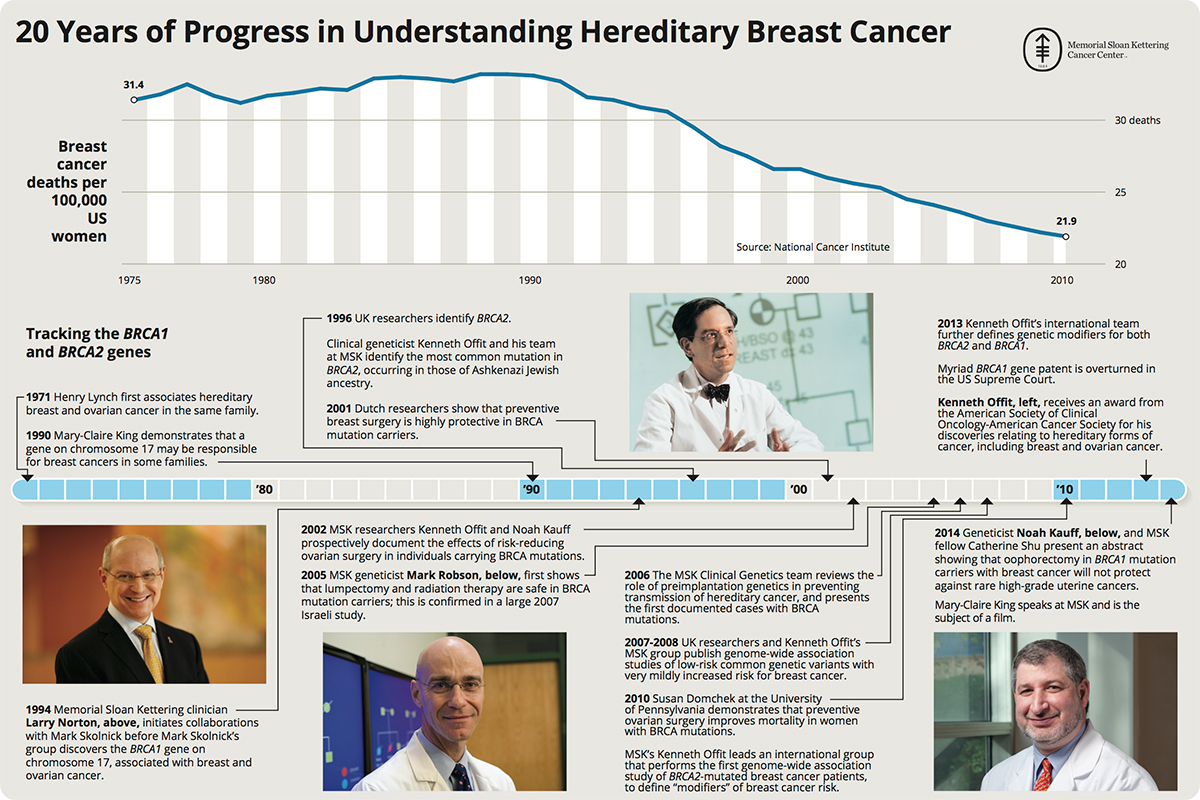
- ↑ "20 Years of Progress in Understanding Breast Cancer" (JPG). Mskcc.org. Retrieved 2015-07-17.
- ↑ Kolata G (1996-10-02). "2d Breast Cancer Gene Found in Jewish Women". The New York Times. ISSN 0362-4331. Retrieved 2015-07-07.
- ↑ ۴۰٫۰ ۴۰٫۱ US patent 5837492, Tavtigian SV, Kamb A, Simard J, Couch F, Rommens JM, Weber BL, "Chromosome 13-linked breast cancer susceptibility gene", issued 1998-11-17, assigned to Myriad Genetics, Inc., Endo Recherche, Inc., HSC Research & Development Limited Partnership, Trustees of the University of Pennsylvania
- ↑ ۴۱٫۰ ۴۱٫۱ الگو:Ref patent
- ↑ US patent 5837492, Tavtigian SV, Kamb A, Simard J, Couch F, Rommens JM, Weber BL, "Chromosome 13-linked breast cancer susceptibility gene", issued 1998-11-17, assigned to Myriad Genetics, Inc., Endo Recherche, Inc., HSC Research & Development Limited Partnership, Trustees of the University of Pennsylvania
- ↑ ۴۳٫۰ ۴۳٫۱ Myriad Investor Page—see "Myriad at a glance" بایگانیشده در ۲۰۱۲-۱۰-۱۸ توسط Wayback Machine accessed October 2012
- ↑ ۴۴٫۰ ۴۴٫۱ ۴۴٫۲ Schwartz J (2009-05-12). "Cancer Patients Challenge the Patenting of a Gene". Health. New York Times.
- ↑ ۴۵٫۰ ۴۵٫۱ Lacroix M, Leclercq G (2005). "The "portrait" of hereditary breast cancer". Breast Cancer Research and Treatment. 89 (3): 297–304. doi:10.1007/s10549-004-2172-4. PMID 15754129. S2CID 23327569.
- ↑ ۴۶٫۰ ۴۶٫۱ Thorlacius S, Olafsdottir G, Tryggvadottir L, Neuhausen S, Jonasson JG, Tavtigian SV, Tulinius H, Ogmundsdottir HM, Eyfjörd JE (1996). "A single BRCA2 mutation in male and female breast cancer families from Iceland with varied cancer phenotypes". Nature Genetics. 13 (1): 117–119. doi:10.1038/ng0596-117. PMID 8673089. S2CID 8443452.
- ↑ ۴۷٫۰ ۴۷٫۱ Thorlacius S, Sigurdsson S, Bjarnadottir H, Olafsdottir G, Jonasson JG, Tryggvadottir L, Tulinius H, Eyfjörd JE (1997). "Study of a single BRCA2 mutation with high carrier frequency in a small population". American Journal of Human Genetics. 60 (5): 1079–1085. PMC 1712443. PMID 9150155.
- ↑ den Dunnen JT, Antonarakis SE (2000). "Mutation nomenclature extensions and suggestions to describe complex mutations: a discussion". Human Mutation. 15 (1): 7–12. doi:10.1002/(SICI)1098-1004(200001)15:1<7::AID-HUMU4>3.0.CO;2-N. PMID 10612815.
- ↑ Neuhausen S, Gilewski T, Norton L, Tran T, McGuire P, Swensen J, Hampel H, Borgen P, Brown K, Skolnick M, Shattuck-Eidens D, Jhanwar S, Goldgar D, Offit K (1996). "Recurrent BRCA2 6174delT mutations in Ashkenazi Jewish women affected by breast cancer". Nature Genetics. 13 (1): 126–128. doi:10.1038/ng0596-126. PMID 8673092. S2CID 11909356.
- ↑ Verhoog LC, van den Ouweland AM, Berns E, van Veghel-Plandsoen MM, van Staveren IL, Wagner A, Bartels CC, Tilanus-Linthorst MM, Devilee P, Seynaeve C, Halley DJ, Niermeijer MF, Klijn JG, Meijers-Heijboer H (2001). "Large regional differences in the frequency of distinct BRCA1/BRCA2 mutations in 517 Dutch breast and/or ovarian cancer families". European Journal of Cancer. 37 (16): 2082–2090. doi:10.1016/S0959-8049(01)00244-1. PMID 11597388.
- ↑ Huusko P, Pääkkönen K, Launonen V, Pöyhönen M, Blanco G, Kauppila A, Puistola U, Kiviniemi H, Kujala M, Leisti J, Winqvist R (1998). "Evidence of founder mutations in Finnish BRCA1 and BRCA2 families". American Journal of Human Genetics. 62 (6): 1544–1548. doi:10.1086/301880. PMC 1377159. PMID 9585608.
- ↑ Pääkkönen K, Sauramo S, Sarantaus L, Vahteristo P, Hartikainen A, Vehmanen P, Ignatius J, Ollikainen V, Kääriäinen H, Vauramo E, Nevanlinna H, Krahe R, Holli K, Kere J (2001). "Involvement of BRCA1 and BRCA2 in breast cancer in a western Finnish sub-population". Genetic Epidemiology. 20 (2): 239–246. doi:10.1002/1098-2272(200102)20:2<239::AID-GEPI6>3.0.CO;2-Y. PMID 11180449. S2CID 41804152.
- ↑ Tonin PN, Mes-Masson AM, Narod SA, Ghadirian P, Provencher D (1999). "Founder BRCA1 and BRCA2 mutations in French Canadian ovarian cancer cases unselected for family history". Clinical Genetics. 55 (5): 318–324. doi:10.1034/j.1399-0004.1999.550504.x. PMID 10422801. S2CID 23931343.
- ↑ Oros KK, Leblanc G, Arcand SL, Shen Z, Perret C, Mes-Masson AM, Foulkes WD, Ghadirian P, Provencher D, Tonin PN (2006). "Haplotype analysis suggests common founders in carriers of recurrent BRCA2 mutation, 3398delAAAAG, in French Canadian hereditary breast and/ovarian cancer families". BMC Medical Genetics. 7 (23): 23. doi:10.1186/1471-2350-7-23. PMC 1464093. PMID 16539696.
- ↑ Tonin PN (2006). "The limited spectrum of pathogenic BRCA1 and BRCA2 mutations in the French Canadian breast and breast-ovarian cancer families, a founder population of Quebec, Canada". Bull Cancer. 93 (9): 841–846. PMID 16980226.
- ↑ Van Der Looij M, Szabo C, Besznyak I, Liszka G, Csokay B, Pulay T, Toth J, Devilee P, King MC, Olah E (2000). "Prevalence of founder BRCA1 and BRCA2 mutations among breast and ovarian cancer patients in Hungary". International Journal of Cancer. 86 (5): 737–740. doi:10.1002/(SICI)1097-0215(20000601)86:5<737::AID-IJC21>3.0.CO;2-1. PMID 10797299. S2CID 25394976.
- ↑ Pisano M, Cossu A, Persico I, Palmieri G, Angius A, Casu G, Palomba G, Sarobba MG, Rocca PC, Dedola MF, Olmeo N, Pasca A, Budroni M, Marras V, Pisano A, Farris A, Massarelli G, Pirastu M, Tanda F (2000). "Identification of a founder BRCA2 mutation in Sardinia". British Journal of Cancer. 82 (3): 553–559. doi:10.1054/bjoc.1999.0963. PMC 2363305. PMID 10682665.
- ↑ ۵۸٫۰ ۵۸٫۱ Scottish/Northern Irish BRCAI/BRCA2 Consortium (2003). "BRCA1 and BRCA2 mutations in Scotland and Northern Ireland". British Journal of Cancer. 88 (8): 1256–1262. doi:10.1038/sj.bjc.6600840. PMC 2747571. PMID 12698193.
- ↑ Liede A, Malik IA, Aziz Z, Rios Pd Pde L, Kwan E, Narod SA (2002). "Contribution of BRCA1 and BRCA2 mutations to breast and ovarian cancer in Pakistan". American Journal of Human Genetics. 71 (3): 595–606. doi:10.1086/342506. PMC 379195. PMID 12181777.
- ↑ Krajc M, De Grève J, Goelen G, Teugels E (2002). "BRCA2 founder mutation in Slovenian breast cancer families". European Journal of Human Genetics. 10 (12): 879–882. doi:10.1038/sj.ejhg.5200886. PMID 12461697.
- ↑ Osorio A, Robledo M, Martínez B, Cebrián A, San Román JM, Albertos J, Lobo F, Benítez J (1998). "Molecular analysis of the BRCA2 gene in 16 breast/ovarian cancer Spanish families". Clin. Genet. 54 (2): 142–7. doi:10.1111/j.1399-0004.1998.tb03717.x. PMID 9761393. S2CID 30388365.
- ↑ Neuhausen SL (2000). "Founder populations and their uses for breast cancer genetics". Cancer Research. 2 (2): 77–81. doi:10.1186/bcr36. PMC 139426. PMID 11250694.
- ↑ ۶۳٫۰ ۶۳٫۱ Seeliger K, Dukowic-Schulze S, Wurz-Wildersinn R, Pacher M, Puchta H (2012). "BRCA2 is a mediator of RAD51- and DMC1-facilitated homologous recombination in Arabidopsis thaliana". New Phytol. 193 (2): 364–75. doi:10.1111/j.1469-8137.2011.03947.x. PMID 22077663.
- ↑ Kojic M, Kostrub CF, Buchman AR, Holloman WK (2002). "BRCA2 homolog required for proficiency in DNA repair, recombination, and genome stability in Ustilago maydis". Mol. Cell. 10 (3): 683–91. doi:10.1016/s1097-2765(02)00632-9. PMID 12408834.
- ↑ Ko E, Lee J, Lee H (2008). "Essential role of brc-2 in chromosome integrity of germ cells in C. elegans". Mol. Cells. 26 (6): 590–4. doi:10.1016/S1016-8478(23)14041-6. PMID 18779660.
- ↑ Martin JS, Winkelmann N, Petalcorin MI, McIlwraith MJ, Boulton SJ (2005). "RAD-51-dependent and -independent roles of a Caenorhabditis elegans BRCA2-related protein during DNA double-strand break repair". Mol. Cell. Biol. 25 (8): 3127–39. doi:10.1128/MCB.25.8.3127-3139.2005. PMC 1069622. PMID 15798199.
- ↑ Klovstad M, Abdu U, Schüpbach T (2008). "Drosophila brca2 is required for mitotic and meiotic DNA repair and efficient activation of the meiotic recombination checkpoint". PLOS Genet. 4 (2): e31. doi:10.1371/journal.pgen.0040031. PMC 2233675. PMID 18266476.
- ↑ Connor F, Bertwistle D, Mee PJ, Ross GM, Swift S, Grigorieva E, Tybulewicz VL, Ashworth A (1997). "Tumorigenesis and a DNA repair defect in mice with a truncating Brca2 mutation". Nat. Genet. 17 (4): 423–30. doi:10.1038/ng1297-423. PMID 9398843. S2CID 42462448.
- ↑ Cotroneo MS, Haag JD, Zan Y, Lopez CC, Thuwajit P, Petukhova GV, Camerini-Otero RD, Gendron-Fitzpatrick A, Griep AE, Murphy CJ, Dubielzig RR, Gould MN (2007). "Characterizing a rat Brca2 knockout model". Oncogene. 26 (11): 1626–35. doi:10.1038/sj.onc.1209960. PMID 16964288.
- ↑ ۷۰٫۰ ۷۰٫۱ Martinez JS, von Nicolai C, Kim T, Ehlén Å, Mazin AV, Kowalczykowski SC, Carreira A (2016). "BRCA2 regulates DMC1-mediated recombination through the BRC repeats". Proc. Natl. Acad. Sci. U.S.A. 113 (13): 3515–20. Bibcode:2016PNAS..113.3515M. doi:10.1073/pnas.1601691113. PMC 4822569. PMID 26976601.
- ↑ ۷۱٫۰ ۷۱٫۱ Frappart PO, Lee Y, Lamont J, McKinnon PJ (2007). "BRCA2 is required for neurogenesis and suppression of medulloblastoma". EMBO J. 26 (11): 2732–42. doi:10.1038/sj.emboj.7601703. PMC 1888666. PMID 17476307.
- ↑ Teng DH, Bogden R, Mitchell J, Baumgard M, Bell R, Berry S, Davis T, Ha PC, Kehrer R, Jammulapati S, Chen Q, Offit K, Skolnick MH, Tavtigian SV, Jhanwar S, Swedlund B, Wong AK, Kamb A (1996). "Low incidence of BRCA2 mutations in breast carcinoma and other cancers". Nat. Genet. 13 (2): 241–4. doi:10.1038/ng0696-241. PMID 8640236. S2CID 9831745.
- ↑ Miki Y, Katagiri T, Kasumi F, Yoshimoto T, Nakamura Y (1996). "Mutation analysis in the BRCA2 gene in primary breast cancers". Nat. Genet. 13 (2): 245–7. doi:10.1038/ng0696-245. PMID 8640237. S2CID 3203046.
- ↑ Lancaster JM, Wooster R, Mangion J, Phelan CM, Cochran C, Gumbs C, Seal S, Barfoot R, Collins N, Bignell G, Patel S, Hamoudi R, Larsson C, Wiseman RW, Berchuck A, Iglehart JD, Marks JR, Ashworth A, Stratton MR, Futreal PA (1996). "BRCA2 mutations in primary breast and ovarian cancers". Nat. Genet. 13 (2): 238–40. doi:10.1038/ng0696-238. PMID 8640235. S2CID 26808443.
- ↑ ۷۵٫۰ ۷۵٫۱ Lee MN, Tseng RC, Hsu HS, Chen JY, Tzao C, Ho WL, Wang YC (2007). "Epigenetic inactivation of the chromosomal stability control genes BRCA1, BRCA2, and XRCC5 in non-small cell lung cancer". Clin. Cancer Res. 13 (3): 832–8. doi:10.1158/1078-0432.CCR-05-2694. PMID 17289874.
- ↑ ۷۶٫۰ ۷۶٫۱ Chan KY, Ozçelik H, Cheung AN, Ngan HY, Khoo US (2002). "Epigenetic factors controlling the BRCA1 and BRCA2 genes in sporadic ovarian cancer". Cancer Res. 62 (14): 4151–6. PMID 12124354.
- ↑ Gu Y, Zhang M, Peng F, Fang L, Zhang Y, Liang H, Zhou W, Ao L, Guo Z (2015). "The BRCA1/2-directed miRNA signature predicts a good prognosis in ovarian cancer patients with wild-type BRCA1/2". Oncotarget. 6 (4): 2397–406. doi:10.18632/oncotarget.2963. PMC 4385859. PMID 25537514.
- ↑ ۷۸٫۰ ۷۸٫۱ Holloman WK (2011). "Unraveling the mechanism of BRCA2 in homologous recombination". Nat. Struct. Mol. Biol. 18 (7): 748–54. doi:10.1038/nsmb.2096. PMC 3647347. PMID 21731065.
- ↑ ۷۹٫۰ ۷۹٫۱ ۷۹٫۲ Bièche I, Noguès C, Lidereau R (1999). "Overexpression of BRCA2 gene in sporadic breast tumours". Oncogene. 18 (37): 5232–8. doi:10.1038/sj.onc.1202903. PMID 10498873.
- ↑ ۸۰٫۰ ۸۰٫۱ ۸۰٫۲ Hedau S, Batra M, Singh UR, Bharti AC, Ray A, Das BC (2015). "Expression of BRCA1 and BRCA2 proteins and their correlation with clinical staging in breast cancer". J Cancer Res Ther. 11 (1): 158–63. doi:10.4103/0973-1482.140985. PMID 25879355.
- ↑ ۸۱٫۰ ۸۱٫۱ Egawa C, Miyoshi Y, Taguchi T, Tamaki Y, Noguchi S (2002). "High BRCA2 mRNA expression predicts poor prognosis in breast cancer patients". Int. J. Cancer. 98 (6): 879–82. doi:10.1002/ijc.10231. PMID 11948466. S2CID 9083282.
- ↑ ۸۲٫۰ ۸۲٫۱ Swisher EM, Gonzalez RM, Taniguchi T, Garcia RL, Walsh T, Goff BA, Welcsh P (2009). "Methylation and protein expression of DNA repair genes: association with chemotherapy exposure and survival in sporadic ovarian and peritoneal carcinomas". Mol. Cancer. 8 (1): 48. doi:10.1186/1476-4598-8-48. PMC 2719582. PMID 19602291.
- ↑ Thike AA, Tan PH, Ikeda M, Iqbal J (2016). "Increased ID4 expression, accompanied by mutant p53 accumulation and loss of BRCA1/2 proteins in triple-negative breast cancer, adversely affects survival". Histopathology. 68 (5): 702–12. doi:10.1111/his.12801. PMID 26259780. S2CID 3566545.
- ↑ ۸۴٫۰ ۸۴٫۱ ۸۴٫۲ ۸۴٫۳ ۸۴٫۴ ۸۴٫۵ Dong Y, Hakimi MA, Chen X, Kumaraswamy E, Cooch NS, Godwin AK, Shiekhattar R (November 2003). "Regulation of BRCC, a holoenzyme complex containing BRCA1 and BRCA2, by a signalosome-like subunit and its role in DNA repair". Mol. Cell. 12 (5): 1087–99. doi:10.1016/S1097-2765(03)00424-6. PMID 14636569.
- ↑ Ryser S, Dizin E, Jefford CE, Delaval B, Gagos S, Christodoulidou A, Krause KH, Birnbaum D, Irminger-Finger I (February 2009). "Distinct roles of BARD1 isoforms in mitosis: full-length BARD1 mediates Aurora B degradation, cancer-associated BARD1beta scaffolds Aurora B and BRCA2". Cancer Res. 69 (3): 1125–34. doi:10.1158/0008-5472.CAN-08-2134. PMID 19176389.
- ↑ ۸۶٫۰ ۸۶٫۱ Liu J, Yuan Y, Huan J, Shen Z (January 2001). "Inhibition of breast and brain cancer cell growth by BCCIPalpha, an evolutionarily conserved nuclear protein that interacts with BRCA2". Oncogene. 20 (3): 336–45. doi:10.1038/sj.onc.1204098. PMID 11313963.
- ↑ ۸۷٫۰ ۸۷٫۱ Sarkisian CJ, Master SR, Huber LJ, Ha SI, Chodosh LA (October 2001). "Analysis of murine Brca2 reveals conservation of protein-protein interactions but differences in nuclear localization signals". J. Biol. Chem. 276 (40): 37640–8. doi:10.1074/jbc.M106281200. PMID 11477095.
- ↑ ۸۸٫۰ ۸۸٫۱ Chen J, Silver DP, Walpita D, Cantor SB, Gazdar AF, Tomlinson G, Couch FJ, Weber BL, Ashley T, Livingston DM, Scully R (September 1998). "Stable interaction between the products of the BRCA1 and BRCA2 tumor suppressor genes in mitotic and meiotic cells". Mol. Cell. 2 (3): 317–28. doi:10.1016/S1097-2765(00)80276-2. PMID 9774970.
- ↑ Reuter TY, Medhurst AL, Waisfisz Q, Zhi Y, Herterich S, Hoehn H, Gross HJ, Joenje H, Hoatlin ME, Mathew CG, Huber PA (October 2003). "Yeast two-hybrid screens imply involvement of Fanconi anemia proteins in transcription regulation, cell signaling, oxidative metabolism, and cellular transport". Exp. Cell Res. 289 (2): 211–21. doi:10.1016/S0014-4827(03)00261-1. PMID 14499622.
- ↑ Futamura M, Arakawa H, Matsuda K, Katagiri T, Saji S, Miki Y, Nakamura Y (March 2000). "Potential role of BRCA2 in a mitotic checkpoint after phosphorylation by hBUBR1". Cancer Res. 60 (6): 1531–5. PMID 10749118.
- ↑ Siddique H, Rao VN, Reddy ES (August 2009). "CBP-mediated post-translational N-glycosylation of BRCA2". Int J Oncol. 35 (2): 16387–91. doi:10.3892/ijo_00000351. PMID 19578754.
- ↑ Hughes-Davies L, Huntsman D, Ruas M, Fuks F, Bye J, Chin SF, Milner J, Brown LA, Hsu F, Gilks B, Nielsen T, Schulzer M, Chia S, Ragaz J, Cahn A, Linger L, Ozdag H, Cattaneo E, Jordanova ES, Schuuring E, Yu DS, Venkitaraman A, Ponder B, Doherty A, Aparicio S, Bentley D, Theillet C, Ponting CP, Caldas C, Kouzarides T (November 2003). "EMSY links the BRCA2 pathway to sporadic breast and ovarian cancer". Cell. 115 (5): 523–35. doi:10.1016/S0092-8674(03)00930-9. PMID 14651845. S2CID 18911371.
- ↑ Wang X, Andreassen PR, D'Andrea AD (July 2004). "Functional interaction of monoubiquitinated FANCD2 and BRCA2/FANCD1 in chromatin". Mol. Cell. Biol. 24 (13): 5850–62. doi:10.1128/MCB.24.13.5850-5862.2004. PMC 480901. PMID 15199141.
- ↑ Hussain S, Wilson JB, Medhurst AL, Hejna J, Witt E, Ananth S, Davies A, Masson JY, Moses R, West SC, de Winter JP, Ashworth A, Jones NJ, Mathew CG (June 2004). "Direct interaction of FANCD2 with BRCA2 in DNA damage response pathways". Hum. Mol. Genet. 13 (12): 1241–8. doi:10.1093/hmg/ddh135. PMID 15115758.
- ↑ Hejna J, Holtorf M, Hines J, Mathewson L, Hemphill A, Al-Dhalimy M, Olson SB, Moses RE (April 2008). "Tip60 is required for DNA interstrand cross-link repair in the Fanconi anemia pathway". J. Biol. Chem. 283 (15): 9844–51. doi:10.1074/jbc.M709076200. PMC 2398728. PMID 18263878.
- ↑ Hussain S, Witt E, Huber PA, Medhurst AL, Ashworth A, Mathew CG (October 2003). "Direct interaction of the Fanconi anaemia protein FANCG with BRCA2/FANCD1". Hum. Mol. Genet. 12 (19): 2503–10. doi:10.1093/hmg/ddg266. PMID 12915460.
- ↑ Yuan Y, Shen Z (December 2001). "Interaction with BRCA2 suggests a role for filamin-1 (hsFLNa) in DNA damage response". J. Biol. Chem. 276 (51): 48318–24. doi:10.1074/jbc.M102557200. PMID 11602572.
- ↑ Marmorstein LY, Kinev AV, Chan GK, Bochar DA, Beniya H, Epstein JA, Yen TJ, Shiekhattar R (January 2001). "A human BRCA2 complex containing a structural DNA binding component influences cell cycle progression". Cell. 104 (2): 247–57. doi:10.1016/S0092-8674(01)00209-4. PMID 11207365. S2CID 5822368.
- ↑ Hakimi MA, Bochar DA, Chenoweth J, Lane WS, Mandel G, Shiekhattar R (May 2002). "A core-BRAF35 complex containing histone deacetylase mediates repression of neuronal-specific genes". Proc. Natl. Acad. Sci. U.S.A. 99 (11): 7420–5. Bibcode:2002PNAS...99.7420H. doi:10.1073/pnas.112008599. PMC 124246. PMID 12032298.
- ↑ ۱۰۰٫۰ ۱۰۰٫۱ ۱۰۰٫۲ Marmorstein LY, Ouchi T, Aaronson SA (November 1998). "The BRCA2 gene product functionally interacts with p53 and RAD51". Proc. Natl. Acad. Sci. U.S.A. 95 (23): 13869–74. Bibcode:1998PNAS...9513869M. doi:10.1073/pnas.95.23.13869. PMC 24938. PMID 9811893.
- ↑ "Entrez Gene: PALB2 partner and localizer of BRCA2".
- ↑ ۱۰۲٫۰ ۱۰۲٫۱ ۱۰۲٫۲ Lin HR, Ting NS, Qin J, Lee WH (September 2003). "M phase-specific phosphorylation of BRCA2 by Polo-like kinase 1 correlates with the dissociation of the BRCA2-P/CAF complex". J. Biol. Chem. 278 (38): 35979–87. doi:10.1074/jbc.M210659200. PMID 12815053.
- ↑ Fuks F, Milner J, Kouzarides T (November 1998). "BRCA2 associates with acetyltransferase activity when bound to P/CAF". Oncogene. 17 (19): 2531–4. doi:10.1038/sj.onc.1202475. PMID 9824164.
- ↑ Lee M, Daniels MJ, Venkitaraman AR (January 2004). "Phosphorylation of BRCA2 by the Polo-like kinase Plk1 is regulated by DNA damage and mitotic progression". Oncogene. 23 (4): 865–72. doi:10.1038/sj.onc.1207223. PMID 14647413.
- ↑ Sharan SK, Morimatsu M, Albrecht U, Lim DS, Regel E, Dinh C, Sands A, Eichele G, Hasty P, Bradley A (April 1997). "Embryonic lethality and radiation hypersensitivity mediated by Rad51 in mice lacking Brca2". Nature. 386 (6627): 804–10. Bibcode:1997Natur.386..804S. doi:10.1038/386804a0. hdl:11858/00-001M-0000-0010-5059-F. PMID 9126738. S2CID 4238943.
- ↑ Yu DS, Sonoda E, Takeda S, Huang CL, Pellegrini L, Blundell TL, Venkitaraman AR (October 2003). "Dynamic control of Rad51 recombinase by self-association and interaction with BRCA2". Mol. Cell. 12 (4): 1029–41. doi:10.1016/S1097-2765(03)00394-0. PMID 14580352.
- ↑ ۱۰۷٫۰ ۱۰۷٫۱ Chen PL, Chen CF, Chen Y, Xiao J, Sharp ZD, Lee WH (April 1998). "The BRC repeats in BRCA2 are critical for RAD51 binding and resistance to methyl methanesulfonate treatment". Proc. Natl. Acad. Sci. U.S.A. 95 (9): 5287–92. Bibcode:1998PNAS...95.5287C. doi:10.1073/pnas.95.9.5287. PMC 20253. PMID 9560268.
- ↑ ۱۰۸٫۰ ۱۰۸٫۱ Wong AK, Pero R, Ormonde PA, Tavtigian SV, Bartel PL (December 1997). "RAD51 interacts with the evolutionarily conserved BRC motifs in the human breast cancer susceptibility gene brca2". J. Biol. Chem. 272 (51): 31941–4. doi:10.1074/jbc.272.51.31941. PMID 9405383.
- ↑ Katagiri T, Saito H, Shinohara A, Ogawa H, Kamada N, Nakamura Y, Miki Y (March 1998). "Multiple possible sites of BRCA2 interacting with DNA repair protein RAD51". Genes Chromosomes Cancer. 21 (3): 217–22. doi:10.1002/(SICI)1098-2264(199803)21:3<217::AID-GCC5>3.0.CO;2-2. PMID 9523196. S2CID 45954246.
- ↑ Pellegrini L, Yu DS, Lo T, Anand S, Lee M, Blundell TL, Venkitaraman AR (November 2002). "Insights into DNA recombination from the structure of a RAD51-BRCA2 complex". Nature. 420 (6913): 287–93. Bibcode:2002Natur.420..287P. doi:10.1038/nature01230. PMID 12442171. S2CID 4359383.
- ↑ Tarsounas M, Davies AA, West SC (January 2004). "RAD51 localization and activation following DNA damage". Philos. Trans. R. Soc. Lond. B Biol. Sci. 359 (1441): 87–93. doi:10.1098/rstb.2003.1368. PMC 1693300. PMID 15065660.
- ↑ Wong JM, Ionescu D, Ingles CJ (January 2003). "Interaction between BRCA2 and replication protein A is compromised by a cancer-predisposing mutation in BRCA2". Oncogene. 22 (1): 28–33. doi:10.1038/sj.onc.1206071. PMID 12527904.
- ↑ Marston NJ, Richards WJ, Hughes D, Bertwistle D, Marshall CJ, Ashworth A (July 1999). "Interaction between the product of the breast cancer susceptibility gene BRCA2 and DSS1, a protein functionally conserved from yeast to mammals". Mol. Cell. Biol. 19 (7): 4633–42. doi:10.1128/MCB.19.7.4633. PMC 84261. PMID 10373512.
- ↑ ۱۱۴٫۰ ۱۱۴٫۱ ۱۱۴٫۲ ۱۱۴٫۳ ۱۱۴٫۴ Yang H, Jeffrey PD, Miller J, Kinnucan E, Sun Y, Thoma NH, Zheng N, Chen PL, Lee WH, Pavletich NP (September 2002). "BRCA2 function in DNA binding and recombination from a BRCA2-DSS1-ssDNA structure". Science. 297 (5588): 1837–48. Bibcode:2002Sci...297.1837Y. doi:10.1126/science.297.5588.1837. PMID 12228710.
- ↑ Preobrazhenska O, Yakymovych M, Kanamoto T, Yakymovych I, Stoika R, Heldin CH, Souchelnytskyi S (August 2002). "BRCA2 and Smad3 synergize in regulation of gene transcription". Oncogene. 21 (36): 5660–4. doi:10.1038/sj.onc.1205732. PMID 12165866.
- ↑ Bork P, Blomberg N, Nilges M (May 1996). "Internal repeats in the BRCA2 protein sequence". Nat. Genet. 13 (1): 22–3. doi:10.1038/ng0596-22. PMID 8673099. S2CID 2312211.
- ↑ Alagar, S.; Bahadur, R. P. (2020). "DSS1 allosterically regulates the conformation of the tower domain of BRCA2 that has dsDNA binding specificity for homologous recombination". International Journal of Biological Macromolecules. 165 (Pt A): 918–929. doi:10.1016/j.ijbiomac.2020.09.230. PMID 33011260. S2CID 222165754.
- ↑ "ACLU sues over patents on breast cancer genes". CNN. Archived from the original on 15 May 2009. Retrieved 2009-05-14.
- ↑ Conley J, Vorhous D, Cook-Deegan J (2011-03-01). "How Will Myriad Respond to the Next Generation of BRCA Testing?". Robinson, Bradshaw, and Hinson. Retrieved 2012-12-09.
- ↑ "Genetics and Patenting". Human Genome Project Information. U.S. Department of Energy Genome Programs. 2010-07-07.
- ↑ Liptak A (13 June 2013). "Supreme Court Rules Human Genes May Not Be Patented". New York Times. Retrieved 13 June 2013.
- ↑ Corderoy A (February 15, 2013). "Landmark patent ruling over breast cancer gene BRCA1". Sydney Morning Herald. Retrieved June 14, 2013.
- ↑ Corderoy A (June 14, 2013). "Companies can't patent genes, US court rules". Sydney Morning Herald. Retrieved June 14, 2013.
{kind=link}
پیوند به بیرون
[ویرایش]- BRCA2 Protein در سرعنوانهای موضوعی پزشکی (MeSH) در کتابخانهٔ ملی پزشکی ایالات متحدهٔ آمریکا
نگارخانه PDB | |
|---|---|
|