Paraganglioma
| Paraganglioma | |
|---|---|
| Malattia rara | |
| Specialità | oncologia |
| Classificazione e risorse esterne (EN) | |
| ICD-O | 8680 e 8680/1 |
| ICD-9-CM | 194.5, 194.6, 227.5, 227.6 e 237.3 |
| ICD-10 | C75.4, C75.5, D35.5, D35.6, D44.6 e D44.7 |
| OMIM | 115310, 614165, 605373, 601650 e 168000 |
| MeSH | D010235 |
| eMedicine | 251009 |
I paragangliomi sono tumori che insorgono dai paragangli extra-surrenalici o surrenalici (in tal caso prendono il nome di feocromocitoma[1]).
Epidemiologia
[modifica | modifica wikitesto]Il paraganglioma/feocromocitoma è un tumore piuttosto raro e ha un'incidenza di circa 2/1 000 000 persone l'anno. Generalmente insorgono nel sesto decennio di vita. Il più delle volte compaiono singolarmente e sporadicamente, ma possono essere familiari, con trasmissione autosomica dominante nella sindrome di neoplasia endocrina multipla MEN II. Ciononostante la penetranza della malattia è incompleta.
Eziologia
[modifica | modifica wikitesto]Genetica
[modifica | modifica wikitesto]Circa il 40% dei tumori mostra mutazioni nei geni codificanti per le 4 sub-unità dell'enzima succinato deidrogenasi, per VHL, RET, TMEM127 e MAX. I tumori che presentano alterazioni nel gene SDHB si originano principalmente da i paragangli simpatici e mostrano frequentemente metastasi.
Anatomia patologica
[modifica | modifica wikitesto]Le caratteristiche morfologiche dei paragangliomi sono abbastanza uniformi: sono composti di nidi di cellule poligonali principali, le cosiddette zellballen (dal tedesco, "palle cellulari"), circondati da stroma vascolare.[2][3] Le cellule tumorali hanno citoplasma abbondante, chiaro o granuloso, eosinofilo e nuclei uniformi, rotondi od ovoidali, a volte vescicolari. Nella maggior parte dei tumori le mitosi sono rare e si osserva uno scarso polimorfismo; la microscopia elettronica spesso evidenzia granuli neuroendocrini ben demarcati nei tumori paravertebrali, che tendono ad essere scarsi invece nei tumori non secernenti. Tramite immunoistochimica si può osservare che le cellule centrali sono positive per marcatori tipici delle cellule neurali (ad esempio la cromogranina) mentre le cellule di sostegno sono positive per i marcatori tipici delle cellule gliali (S100, proteina fibrillare acida della glia).
Fisiopatologia
[modifica | modifica wikitesto]I paragangliomi presentano caratteristiche differenti a seconda della tipologia di paraganglio da cui si originano:
- I tumori che si sviluppano dai paragangli associati al sistema nervoso parasimpatico (paraganglioma parasimpatico o paraganglioma della testa del collo o chemodectoma) che compongono la catena aorto-polmonare, mostrano una crescita lenta, sono benigni (non formano metastasi) e non producono catecolamine. Ciononostante possono provocare seri danni al paziente a causa della compressione delle delicate strutture adiacenti (vene e nervi) e per la tendenza ad infiltrarsi nella regione cranica. Sono la variante più rara della malattia (3%).
- I tumori che si sviluppano dai paragangli associati al sistema nervoso simpatico (paraganglioma simpatico o addominale) mostrano una crescita rapida, producono spesso catecolamine (in genere adrenalina) in maniera costitutiva o parossistica; tendono a formare metastasi.
Clinica
[modifica | modifica wikitesto]Sintomatologia
[modifica | modifica wikitesto]I paragangliomi localizzati nella regione della testa e del collo possono provocare deficit dei nervi cranici. In particolare del nervo vago: acufeni, disfonia, senso di ripienezza faringea, paralisi facciale, difficoltà di movimento della lingua. I paragangliomi giugulo-timpanici presentano invece una sintomatologia precoce, rappresentata da acufene pulsatile, in alcuni casi associato a ipoacusia di tipo conduttivo. I paragangliomi secernenti catecolamine in maniera costitutiva o parossistica possono produrre, oltre che i vari problemi dovuti all'accrescimento della loro massa, vari disagi come: ipertensione arteriosa (80-90% dei casi), cefalea (60-90%), tachicardia (50-70%), iperidrosi (55-75%), tremori, pallore (40-45%), ansia e anche attacchi di panico (20-40%). Poiché questi sintomi sono comuni anche ad altre malattie il paraganglioma è stato anche chiamato con il titolo di grande imitatore.[4]
Diagnostica
[modifica | modifica wikitesto]La procedura biochimica si fonda sull'evidenza di alterata secrezione di catecolamine e/o metanefrine. La procedura per immagine anatomica tramite TC e RMN, e per immagine funzionale tramite scintigrafia.
Trattamento
[modifica | modifica wikitesto]La principale terapia è la rimozione chirurgica, l'embolizzazione[5] e, quando non è possibile l'intervento chirurgico, la radioterapia[6]. Per le forme addominali si esegue in genere la laparoscopia mentre per i paragangliomi della regione testa/collo bisogna valutare il rapporto rischio/beneficio offerto dalla chirurgia. La terapia medica è molto importante per la preparazione all'intervento chirurgico dei paragangliomi secernenti. Si basa sull'uso degli alfa-antagonisti.
Galleria d'immagini
[modifica | modifica wikitesto]-
 Microfotografia di un paraganglioma del glomo carotideo.
Microfotografia di un paraganglioma del glomo carotideo. -
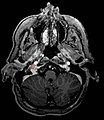 Paraganglioma del glomo giugulare.
Paraganglioma del glomo giugulare.


Note
[modifica | modifica wikitesto]- ^ Capella C. et al, Histopathology, cytology and cytochemistry of pheochromocytomas and paragangliomas including chemodectomas. Pathol Res Pract 186:176, 1988
- ^ Kairi-Vassilatou E, Argeitis J, Nika H, Grapsa D, Smyrniotis V, Kondi-Pafiti A, Malignant paraganglioma of the urinary bladder in a 44-year-old female: clinicopathological and immunohistochemical study of a rare entity and literature review, in Eur. J. Gynaecol. Oncol., vol. 28, n. 2, 2007, pp. 149–51, PMID 17479683.
- ^ G.M. Mariuzzi, 8.1 vie aeree superiori, in Anatomia Patologica e correlazioni anatomo-cliniche, 2007, ISBN 978-88-299-1769-3.
- ^ H. Chen, R. S. Sippel,S. M. O'Dorisio,A. I. Vinik, R. V. Lloyd e K. Pacak. The North American Neuroendocrine Tumor Society Consensus Guideline for the Diagnosis and Management of Neuroendocrine Tumors Pheochromocytoma, Paraganglioma, and Medullary Thyroid Cancer. Pancreas. Volume 39, Number 6, August 2010. Entrez PubMed 20664475
- ^ Carlsen CS, Godballe C, Krogdahl AS, Edal AL, Malignant vagal paraganglioma: report of a case treated with embolization and surgery, in Auris, nasus, larynx, vol. 30, n. 4, 2003, pp. 443–6, DOI:10.1016/S0385-8146(03)00066-X, PMID 14656575.
- ^ Pitiakoudis M, Koukourakis M, Tsaroucha A, Manavis J, Polychronidis A, Simopoulos C, Malignant retroperitoneal paraganglioma treated with concurrent radiotherapy and chemotherapy, in Clinical oncology (Royal College of Radiologists (Great Britain)), vol. 16, n. 8, 2004, pp. 580–1, PMID 15630855.
Bibliografia
[modifica | modifica wikitesto]- Robbins e Cotran, "Le basi Patologiche delle Malattie", Ed. Elsevier e Masson
- ENDOWIKI, http://endowiki.it/index.php?option=com_content&view=article&id=353&Itemid=549&lang=it
Altri progetti
[modifica | modifica wikitesto] Wikimedia Commons contiene immagini o altri file su paraganglioma
Wikimedia Commons contiene immagini o altri file su paraganglioma