



-
 1a1u: SOLUTION STRUCTURE DETERMINATION OF A P53 MUTANT DIMERIZATION DOMAIN, NMR, MINIMIZED AVERAGE STRUCTURE
1a1u: SOLUTION STRUCTURE DETERMINATION OF A P53 MUTANT DIMERIZATION DOMAIN, NMR, MINIMIZED AVERAGE STRUCTURE -
 1aie: P53 TETRAMERIZATION DOMAIN CRYSTAL STRUCTURE
1aie: P53 TETRAMERIZATION DOMAIN CRYSTAL STRUCTURE -
 1c26: CRYSTAL STRUCTURE OF P53 TETRAMERIZATION DOMAIN
1c26: CRYSTAL STRUCTURE OF P53 TETRAMERIZATION DOMAIN -
 1gzh: CRYSTAL STRUCTURE OF THE BRCT DOMAINS OF HUMAN 53BP1 BOUND TO THE P53 TUMOR SUPPRESSOR
1gzh: CRYSTAL STRUCTURE OF THE BRCT DOMAINS OF HUMAN 53BP1 BOUND TO THE P53 TUMOR SUPPRESSOR -
 1hs5: NMR SOLUTION STRUCTURE OF DESIGNED P53 DIMER
1hs5: NMR SOLUTION STRUCTURE OF DESIGNED P53 DIMER -
 1kzy: Crystal Structure of the 53bp1 BRCT Region Complexed to Tumor Suppressor P53
1kzy: Crystal Structure of the 53bp1 BRCT Region Complexed to Tumor Suppressor P53 -
 1olg: HIGH-RESOLUTION SOLUTION STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR
1olg: HIGH-RESOLUTION SOLUTION STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR -
 1olh: HIGH-RESOLUTION SOLUTION STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR
1olh: HIGH-RESOLUTION SOLUTION STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR -
 1pes: NMR SOLUTION STRUCTURE OF THE TETRAMERIC MINIMUM TRANSFORMING DOMAIN OF P53
1pes: NMR SOLUTION STRUCTURE OF THE TETRAMERIC MINIMUM TRANSFORMING DOMAIN OF P53 -
 1pet: NMR SOLUTION STRUCTURE OF THE TETRAMERIC MINIMUM TRANSFORMING DOMAIN OF P53
1pet: NMR SOLUTION STRUCTURE OF THE TETRAMERIC MINIMUM TRANSFORMING DOMAIN OF P53 -
 1sae: HIGH RESOLUTION SOLUTION NMR STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR (SAC STRUCTURES)
1sae: HIGH RESOLUTION SOLUTION NMR STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR (SAC STRUCTURES) -
 1saf: HIGH RESOLUTION SOLUTION NMR STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR (SAD STRUCTURES)
1saf: HIGH RESOLUTION SOLUTION NMR STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR (SAD STRUCTURES) -
 1sag:
1sag: -
 1sah: HIGH RESOLUTION SOLUTION NMR STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR (SAD STRUCTURES)
1sah: HIGH RESOLUTION SOLUTION NMR STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR (SAD STRUCTURES) -
 1sai:
1sai: -
 1saj: HIGH RESOLUTION SOLUTION NMR STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR (SAD STRUCTURES)
1saj: HIGH RESOLUTION SOLUTION NMR STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR (SAD STRUCTURES) -
 1sak: HIGH RESOLUTION SOLUTION NMR STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR (SAC STRUCTURES)
1sak: HIGH RESOLUTION SOLUTION NMR STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR (SAC STRUCTURES) -
 1sal: HIGH RESOLUTION SOLUTION NMR STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR (SAD STRUCTURES)
1sal: HIGH RESOLUTION SOLUTION NMR STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR (SAD STRUCTURES) -
 1tsr: P53 CORE DOMAIN IN COMPLEX WITH DNA
1tsr: P53 CORE DOMAIN IN COMPLEX WITH DNA -
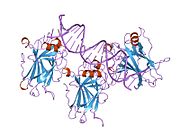 1tup: TUMOR SUPPRESSOR P53 COMPLEXED WITH DNA
1tup: TUMOR SUPPRESSOR P53 COMPLEXED WITH DNA -
 1uol: CRYSTAL STRUCTURE OF THE HUMAN P53 CORE DOMAIN MUTANT M133L/V203A/N239Y/N268D AT 1.9 A RESOLUTION.
1uol: CRYSTAL STRUCTURE OF THE HUMAN P53 CORE DOMAIN MUTANT M133L/V203A/N239Y/N268D AT 1.9 A RESOLUTION. -
 1ycs: P53-53BP2 COMPLEX
1ycs: P53-53BP2 COMPLEX -
 2ac0: Structural Basis of DNA Recognition by p53 Tetramers (complex I)
2ac0: Structural Basis of DNA Recognition by p53 Tetramers (complex I) -
 2ady: Structural Basis of DNA Recognition by p53 Tetramers (complex IV)
2ady: Structural Basis of DNA Recognition by p53 Tetramers (complex IV) -
 2ahi: Structural Basis of DNA Recognition by p53 Tetramers (complex III)
2ahi: Structural Basis of DNA Recognition by p53 Tetramers (complex III) -
 2ata: Structural Basis of DNA Recognition by p53 Tetramers (complex II)
2ata: Structural Basis of DNA Recognition by p53 Tetramers (complex II) -
 2b3g: p53N (fragment 33-60) bound to RPA70N
2b3g: p53N (fragment 33-60) bound to RPA70N -
 2bim: HUMAN P53 CORE DOMAIN MUTANT M133L-V203A-N239Y-N268D-R273H
2bim: HUMAN P53 CORE DOMAIN MUTANT M133L-V203A-N239Y-N268D-R273H -
 2bin: HUMAN P53 CORE DOMAIN MUTANT M133L-H168R-V203A-N239Y-N268D
2bin: HUMAN P53 CORE DOMAIN MUTANT M133L-H168R-V203A-N239Y-N268D -
 2bio: HUMAN P53 CORE DOMAIN MUTANT M133L-V203A-N239Y-R249S-N268D
2bio: HUMAN P53 CORE DOMAIN MUTANT M133L-V203A-N239Y-R249S-N268D -
 2bip: HUMAN P53 CORE DOMAIN MUTANT M133L-H168R-V203A-N239Y-R249S-N268D
2bip: HUMAN P53 CORE DOMAIN MUTANT M133L-H168R-V203A-N239Y-R249S-N268D -
 2biq: HUMAN P53 CORE DOMAIN MUTANT T123A-M133L-H168R-V203A-N239Y-R249S-N268D
2biq: HUMAN P53 CORE DOMAIN MUTANT T123A-M133L-H168R-V203A-N239Y-R249S-N268D -
 2fej: Solution structure of human p53 DNA binding domain.
2fej: Solution structure of human p53 DNA binding domain. -
 2gs0: NMR structure of the complex between the PH domain of the Tfb1 subunit from TFIIH and the activation domain of p53
2gs0: NMR structure of the complex between the PH domain of the Tfb1 subunit from TFIIH and the activation domain of p53 -
 2h1l: The Structure of the Oncoprotein SV40 Large T Antigen and p53 Tumor Suppressor Complex
2h1l: The Structure of the Oncoprotein SV40 Large T Antigen and p53 Tumor Suppressor Complex -
 2j1w: HUMAN P53 CORE DOMAIN MUTANT M133L-V143A-V203A-N239Y-N268D
2j1w: HUMAN P53 CORE DOMAIN MUTANT M133L-V143A-V203A-N239Y-N268D -
 2j1x: HUMAN P53 CORE DOMAIN MUTANT M133L-V203A-Y220C-N239Y-N268D
2j1x: HUMAN P53 CORE DOMAIN MUTANT M133L-V203A-Y220C-N239Y-N268D -
 2j1y: HUMAN P53 CORE DOMAIN MUTANT M133L-V203A-N239Y-G245S-N268D
2j1y: HUMAN P53 CORE DOMAIN MUTANT M133L-V203A-N239Y-G245S-N268D -
 2j1z: HUMAN P53 CORE DOMAIN MUTANT M133L-V203A-N239Y-N268D-F270L
2j1z: HUMAN P53 CORE DOMAIN MUTANT M133L-V203A-N239Y-N268D-F270L -
 2j20: HUMAN P53 CORE DOMAIN MUTANT M133L-V203A-N239Y-N268D-R273C
2j20: HUMAN P53 CORE DOMAIN MUTANT M133L-V203A-N239Y-N268D-R273C -
 2j21: HUMAN P53 CORE DOMAIN MUTANT M133L-V203A-N239Y-N268D-R282W
2j21: HUMAN P53 CORE DOMAIN MUTANT M133L-V203A-N239Y-N268D-R282W -
 2ocj: Human p53 core domain in the absence of DNA
2ocj: Human p53 core domain in the absence of DNA -
 3sak: HIGH RESOLUTION SOLUTION NMR STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR (SAC STRUCTURES)
3sak: HIGH RESOLUTION SOLUTION NMR STRUCTURE OF THE OLIGOMERIZATION DOMAIN OF P53 BY MULTI-DIMENSIONAL NMR (SAC STRUCTURES)
P53
Bài viết này cần thêm chú thích nguồn gốc để kiểm chứng thông tin. |
Tumor protein p53, hay còn gọi là p53 là một protein quan trọng nằm trong điều hòa chu kỳ tế bào - gọi là gen áp chế khối u p53. Khi có tổn thương ở DNA, p53 làm ngừng chu kỳ tế bào cho đến khi DNA bị tổn thương được sửa chữa hoặc p53 có thể làm sự chết rụng tế bào (tiếng Anh apoptosis) nếu không còn khả năng sửa chữa DNA.

Rb là gen có chức năng ngăn cản diễn tiến chu kỳ tế bào bằng cách gắn kết với E2F1 và ngăn cản sự sao chép các gen cần thiết cho tế bào vào pha S. Sở dĩ p53 ngăn cản được chu trình tế bào vì nó hoạt hóa quá trình phiên mã tạo ra CKI, P21 để luân phiên ức chế sự hoạt hóa của CDK. Một khi CDK bị hoạt hóa nó sẽ phosphoryl hóa Rb và làm mất tác dụng của Rb.
Những đột biến mất chức năng p53 làm tăng tính bất ổn định di truyền và làm giảm chết tế bào theo lập trình. Người ta phát hiện thấy trên 50% người mắc các bệnh về ung thư (như ung thư vú, ung thư đại tràng, ung thư phổi, ung thư gan...) đều có những điểm khác biệt trên gene mã hoá p53 so với người bình thường.
Gen
[sửa | sửa mã nguồn]Ở người, gen TP53 nằm trên cánh tay ngắn của nhiễm sắc thể 17 (17p13.1).[2][3][4][5] Gen này trải dài 20 kb, với exon 1 không mã hóa và intron đầu tiên rất dài 10 kb, chồng lên gen Hp53int1. Trình tự mã hóa chứa năm vùng cho thấy mức độ bảo tồn cao ở động vật có xương sống, chủ yếu ở exon 2, 5, 6, 7 và 8, nhưng các trình tự tìm thấy ở động vật không xương sống chỉ cho thấy sự tương đồng xa với TP53 ở động vật có vú.[6] Các gen tương đồng của TP53 [7]đã được xác định ở hầu hết các loài động vật có vú có dữ liệu bộ gen hoàn chỉnh.
Gen TP53 ở người
[sửa | sửa mã nguồn]Ở người, một đa hình phổ biến liên quan đến việc thay thế arginine bằng proline ở vị trí codon 72 của exon 4. Nhiều nghiên cứu đã điều tra mối liên hệ di truyền giữa biến thể này và nguy cơ mắc ung thư; tuy nhiên, kết quả vẫn còn gây tranh cãi. Ví dụ, một phân tích tổng hợp từ năm 2009 đã không chỉ ra mối liên hệ với ung thư cổ tử cung.[8] Một nghiên cứu năm 2011 phát hiện ra rằng đột biến proline TP53 có tác động sâu sắc đến nguy cơ ung thư tuyến tụy ở nam giới.[9] Một nghiên cứu về phụ nữ Ả Rập phát hiện ra rằng đồng hợp tử proline ở codon 72 TP53 có liên quan đến việc giảm nguy cơ ung thư vú.[10] Một nghiên cứu cho thấy rằng các đa hình codon 72 TP53, MDM2 SNP309 và A2164G có thể cùng nhau liên quan đến khả năng mắc ung thư không phải ở vòm họng và rằng MDM2 SNP309 kết hợp với codon 72 TP53 có thể đẩy nhanh quá trình phát triển ung thư không phải ở vòm họng ở phụ nữ.[11] Một nghiên cứu năm 2011 phát hiện ra rằng đa hình codon 72 TP53 có liên quan đến việc tăng nguy cơ ung thư phổi.[12]
Các phân tích tổng hợp từ năm 2011 không tìm thấy mối liên hệ đáng kể nào giữa các đa hình codon 72 TP53 và cả nguy cơ ung thư trực tràng[13] và nguy cơ ung thư nội mạc tử cung.[14] Một nghiên cứu năm 2011 về nhóm sinh con ở Brazil đã tìm thấy mối liên hệ giữa arginine TP53 không đột biến và những cá nhân không có tiền sử gia đình mắc bệnh ung thư.[15] Một nghiên cứu khác năm 2011 phát hiện ra rằng kiểu gen p53 đồng hợp tử (Pro/Pro) có liên quan đến nguy cơ ung thư biểu mô tế bào thận tăng đáng kể.[16]
Cấu trúc và chức năng
[sửa | sửa mã nguồn]p53 là một yếu tố phiên mã điều chỉnh biểu hiện của nhiều gen liên quan đến:
- Chu kỳ tế bào bị dừng lại: Ngăn chặn sự phân chia tế bào nếu phát hiện tổn thương DNA.
- Sửa chữa DNA: Kích hoạt các cơ chế sửa chữa để sửa chữa các đột biến gen.
- Apoptosis (chết tế bào theo chương trình): Gây ra cái chết của tế bào nếu tổn thương DNA không thể phục hồi.
- Lão hóa: Ngăn chặn các tế bào bị tổn thương tăng sinh.
Vai trò trong ung thư
[sửa | sửa mã nguồn]Đột biến gen TP53 là một trong những biến đổi di truyền phổ biến nhất trong ung thư ở người, được tìm thấy ở khoảng 50% tất cả các khối u.[17] Khi p53 bị bất hoạt hoặc đột biến, các tế bào mất khả năng điều chỉnh phân chia đúng cách, dẫn đến sự phát triển tế bào không kiểm soát và hình thành khối u.[18]
Quy định
[sửa | sửa mã nguồn]p53 hoạt động như một cảm biến căng thẳng của tế bào. Thông thường, nó được giữ ở mức thấp bằng cách liên tục được đánh dấu để phân hủy bởi protein ligase ubiquitin E3 MDM2.[19] p53 được kích hoạt để đáp ứng với vô số tác nhân gây căng thẳng – bao gồm tổn thương DNA (do tia UV, IR hoặc tác nhân hóa học như hydrogen peroxide gây ra), căng thẳng oxy hóa,[20] sốc thẩm thấu, suy giảm ribonucleotide, nhiễm trùng phổi do vi-rút[47] và biểu hiện oncogen mất điều hòa. Sự kích hoạt này được đánh dấu bằng hai sự kiện chính. Đầu tiên, thời gian bán hủy của protein p53 tăng mạnh, dẫn đến sự tích tụ nhanh chóng của p53 trong các tế bào bị căng thẳng. Thứ hai, một sự thay đổi cấu hình buộc p53 phải được kích hoạt như một chất điều hòa phiên mã trong các tế bào này. Sự kiện quan trọng dẫn đến việc kích hoạt p53 là sự phosphoryl hóa miền đầu N của nó. Miền hoạt hóa phiên mã đầu N chứa một số lượng lớn các vị trí phosphoryl hóa và có thể được coi là mục tiêu chính cho các protein kinase chuyển tín hiệu căng thẳng. [cần dẫn nguồn]
Các protein kinase được biết là nhắm vào miền hoạt hóa phiên mã này của p53 có thể được chia thành hai nhóm. Nhóm protein kinase đầu tiên thuộc họ MAPK (JNK1-3, ERK1-2, p38 MAPK), được biết là phản ứng với một số loại căng thẳng, chẳng hạn như tổn thương màng, căng thẳng oxy hóa, sốc thẩm thấu, sốc nhiệt, v.v. Nhóm protein kinase thứ hai (ATR, ATM, CHK1 và CHK2, DNA-PK, CAK, TP53RK) có liên quan đến điểm kiểm tra tính toàn vẹn của bộ gen, một chuỗi phân tử phát hiện và phản ứng với một số dạng tổn thương DNA do căng thẳng gây độc gen. Các oncogen cũng kích thích hoạt hóa p53, được trung gian bởi protein p14ARF. [cần dẫn nguồn]
Trong các tế bào không bị căng thẳng, mức p53 được giữ ở mức thấp thông qua quá trình phân hủy liên tục của p53. Một loại protein có tên là Mdm2 (còn được gọi là HDM2 ở người), liên kết với p53, ngăn chặn hoạt động của nó và vận chuyển nó từ nhân đến tế bào chất. Mdm2 cũng hoạt động như một ubiquitin ligase và liên kết cộng hóa trị ubiquitin với p53 và do đó đánh dấu p53 để phân hủy bởi proteasome. Tuy nhiên, ubiquitylation của p53 là có thể đảo ngược. Khi kích hoạt p53, Mdm2 cũng được kích hoạt, thiết lập một vòng phản hồi. Mức p53 có thể biểu hiện dao động (hoặc xung lặp lại) để đáp ứng với một số căng thẳng nhất định và những xung này có thể quan trọng trong việc xác định xem các tế bào có sống sót sau căng thẳng hay chết.[48]
MI-63 liên kết với MDM2, kích hoạt lại p53 trong những tình huống mà chức năng của p53 bị ức chế.[49]
Một protease đặc hiệu ubiquitin, USP7 (hay HAUSP), có thể cắt ubiquitin khỏi p53, do đó bảo vệ nó khỏi sự thoái hóa phụ thuộc proteasome thông qua con đường ubiquitin ligase. Đây là một phương tiện mà p53 được ổn định để đáp ứng với các tác nhân gây ung thư. USP42 cũng đã được chứng minh là có khả năng khử ubiquitin p53 và có thể cần thiết để p53 có khả năng phản ứng với căng thẳng.[50]
Nghiên cứu gần đây đã chỉ ra rằng HAUSP chủ yếu nằm trong nhân, mặc dù một phần của nó có thể được tìm thấy trong tế bào chất và ty thể. Việc biểu hiện quá mức HAUSP dẫn đến sự ổn định của p53. Tuy nhiên, sự suy giảm HAUSP không dẫn đến sự giảm nồng độ p53 mà thay vào đó làm tăng nồng độ p53 do thực tế là HAUSP liên kết và khử ubiquitin Mdm2. Người ta đã chứng minh rằng HAUSP là đối tác liên kết tốt hơn với Mdm2 so với p53 trong các tế bào không bị căng thẳng.
Tuy nhiên, USP10 đã được chứng minh là nằm trong tế bào chất ở các tế bào không bị căng thẳng và khử ubiquitin p53 trong tế bào chất, đảo ngược quá trình ubiquitin hóa Mdm2. Sau khi DNA bị tổn thương, USP10 chuyển đến nhân và góp phần vào sự ổn định của p53. USP10 cũng không tương tác với Mdm2.[51]
Quá trình phosphoryl hóa đầu N của p53 bởi các protein kinase được đề cập ở trên làm gián đoạn sự liên kết Mdm2. Các protein khác, chẳng hạn như Pin1, sau đó được tuyển dụng vào p53 và gây ra sự thay đổi cấu hình trong p53, ngăn chặn sự liên kết Mdm2 nhiều hơn nữa. Quá trình phosphoryl hóa cũng cho phép liên kết các đồng hoạt hóa phiên mã, như p300 và PCAF, sau đó acetylate đầu C của p53, để lộ vùng liên kết DNA của p53, cho phép nó kích hoạt hoặc ức chế các gen cụ thể. Enzym deacetylase, chẳng hạn như Sirt1 và Sirt7, có thể deacetyl hóa p53, dẫn đến ức chế apoptosis.[52] Một số oncogen cũng có thể kích thích phiên mã các protein liên kết với MDM2 và ức chế hoạt động của nó. [cần dẫn nguồn]
Các dấu hiệu biểu sinh như metyl hóa histone cũng có thể điều chỉnh p53, ví dụ, p53 tương tác trực tiếp với cofactor Trim24 ức chế liên kết với histone ở các vùng của bộ gen bị ức chế biểu sinh.[53] Trim24 ngăn p53 kích hoạt các mục tiêu của nó, nhưng chỉ ở những vùng này, về cơ bản cung cấp cho p53 khả năng 'đọc' hồ sơ histone tại các gen mục tiêu chính và hoạt động theo cách đặc hiệu của gen. [cần dẫn nguồn]
Hình ảnh
[sửa | sửa mã nguồn]Tham khảo
[sửa | sửa mã nguồn]- ^ “Human PubMed Reference:”.
- ^ Matlashewski, G.; Lamb, P.; Pim, D.; Peacock, J.; Crawford, L.; Benchimol, S. (tháng 12 năm 1984). “Isolation and characterization of a human p53 cDNA clone: expression of the human p53 gene”. The EMBO Journal (bằng tiếng Anh). 3 (13): 3257–3262. doi:10.1002/j.1460-2075.1984.tb02287.x.
- ^ Isobe, M.; Emanuel, B. S.; Givol, D.; Oren, M.; Croce, C. M. (tháng 3 năm 1986). “Localization of gene for human p53 tumour antigen to band 17p13”. Nature (bằng tiếng Anh). 320 (6057): 84–85. doi:10.1038/320084a0. ISSN 0028-0836.
- ^ Kern, Scott E.; Kinzler, Kenneth W.; Bruskin, Arthur; Jarosz, David; Friedman, Paula; Prives, Carol; Vogelstein, Bert (21 tháng 6 năm 1991). “Identification of p53 as a Sequence-Specific DNA-Binding Protein”. Science (bằng tiếng Anh). 252 (5013): 1708–1711. doi:10.1126/science.2047879. ISSN 0036-8075.
- ^ McBride, O W; Merry, D; Givol, D (tháng 1 năm 1986). “The gene for human p53 cellular tumor antigen is located on chromosome 17 short arm (17p13)”. Proceedings of the National Academy of Sciences (bằng tiếng Anh). 83 (1): 130–134. doi:10.1073/pnas.83.1.130. ISSN 0027-8424.
- ^ May, Pierre; May, Evelyne (13 tháng 12 năm 1999). “Twenty years of p53 research: structural and functional aspects of the p53 protein”. Oncogene (bằng tiếng Anh). 18 (53): 7621–7636. doi:10.1038/sj.onc.1203285. ISSN 0950-9232.
- ^ “Supplemental Information 1: Sequence retrieved from GenBank for phylogenetic analysis”. doi.org. Truy cập ngày 2 tháng 3 năm 2025.
- ^ Klug, Stefanie J; Ressing, Meike; Koenig, Jochem; Abba, Martin C; Agorastos, Theodoros; Brenna, Sylvia MF; Ciotti, Marco; Das, Br; Del Mistro, Annarosa (tháng 8 năm 2009). “TP53 codon 72 polymorphism and cervical cancer: a pooled analysis of individual data from 49 studies”. The Lancet Oncology (bằng tiếng Anh). 10 (8): 772–784. doi:10.1016/S1470-2045(09)70187-1.
- ^ Sonoyama, Takayuki (8 tháng 3 năm 2011). “TP53 codon 72 polymorphism is associated with pancreatic cancer risk in males, smokers and drinkers”. Molecular Medicine Reports. doi:10.3892/mmr.2011.449. ISSN 1791-2997.
- ^ Alawadi, Shafika; Ghabreau, Lina; Alsaleh, Mervat; Abdulaziz, Zainab; Rafeek, Mohamed; Akil, Nizar; Alkhalaf, Moussa (tháng 9 năm 2011). “P53 gene polymorphisms and breast cancer risk in Arab women”. Medical Oncology (bằng tiếng Anh). 28 (3): 709–715. doi:10.1007/s12032-010-9505-4. ISSN 1357-0560.
- ^ Yu, Hongping; Huang, Yu‐jing; Liu, Zhensheng; Wang, Li‐E; Li, Guojun; Sturgis, Erich M.; Johnson, David G.; Wei, Qingyi (tháng 9 năm 2011). “Effects of MDM2 promoter polymorphisms and p53 codon 72 polymorphism on risk and age at onset of squamous cell carcinoma of the head and neck”. Molecular Carcinogenesis (bằng tiếng Anh). 50 (9): 697–706. doi:10.1002/mc.20806. ISSN 0899-1987.
- ^ Piao, Jin-Mei; Kim, Hee Nam; Song, Hye-Rim; Kweon, Sun-Seog; Choi, Jin-Su; Yun, Woo-Jun; Kim, Young-Chul; Oh, In-Jae; Kim, Kyu-Sik (tháng 9 năm 2011). “p53 codon 72 polymorphism and the risk of lung cancer in a Korean population”. Lung Cancer (bằng tiếng Anh). 73 (3): 264–267. doi:10.1016/j.lungcan.2010.12.017.
- ^ Wang, Jing-Jun; Zheng, Yuan; Sun, Liang; Wang, Li; Yu, Peng-Bo; Dong, Jian-Hua; Zhang, Lei; Xu, Jing; Shi, Wei (tháng 11 năm 2011). “TP53 codon 72 polymorphism and colorectal cancer susceptibility: a meta-analysis”. Molecular Biology Reports (bằng tiếng Anh). 38 (8): 4847–4853. doi:10.1007/s11033-010-0619-8. ISSN 0301-4851.
- ^ Jiang, De-Ke; Yao, Lei; Ren, Wei-Hua; Wang, Wen-Zhang; Peng, Bo; Yu, Long (tháng 12 năm 2011). “TP53 Arg72Pro polymorphism and endometrial cancer risk: a meta-analysis”. Medical Oncology (bằng tiếng Anh). 28 (4): 1129–1135. doi:10.1007/s12032-010-9597-x. ISSN 1357-0560.
- ^ Thurow, Helena S; Haack, Ricardo; Hartwig, Fernando P; de Oliveira, Isabel O; Dellagostin, Odir A; Gigante, Denise P; Horta, Bernardo L; Collares, Tiago; Seixas, Fabiana K (tháng 12 năm 2011). “TP53 gene polymorphism: Importance to cancer, ethnicity and birth weight in a Brazilian cohort”. Journal of Biosciences (bằng tiếng Anh). 36 (5): 823–831. doi:10.1007/s12038-011-9147-5. ISSN 0250-5991.
- ^ Huang, Chao-Yuan; Su, Chien-Tien; Chu, Jan-Show; Huang, Shu-Pin; Pu, Yeong-Shiau; Yang, Hsiu-Yuan; Chung, Chi-Jung; Wu, Chia-Chang; Hsueh, Yu-Mei (tháng 12 năm 2011). “The polymorphisms of P53 codon 72 and MDM2 SNP309 and renal cell carcinoma risk in a low arsenic exposure area”. Toxicology and Applied Pharmacology (bằng tiếng Anh). 257 (3): 349–355. doi:10.1016/j.taap.2011.09.018.
- ^ Levine, Arnold J (tháng 2 năm 1997). “p53, the Cellular Gatekeeper for Growth and Division”. Cell. 88 (3): 323–331. doi:10.1016/s0092-8674(00)81871-1. ISSN 0092-8674.
- ^ Vogelstein, Bert; Lane, David; Levine, Arnold J. (tháng 11 năm 2000). “Surfing the p53 network”. Nature. 408 (6810): 307–310. doi:10.1038/35042675. ISSN 0028-0836.
- ^ Bykov, Vladimir J. N.; Eriksson, Sofi E.; Bianchi, Julie; Wiman, Klas G. (tháng 2 năm 2018). “Targeting mutant p53 for efficient cancer therapy”. Nature Reviews Cancer (bằng tiếng Anh). 18 (2): 89–102. doi:10.1038/nrc.2017.109. ISSN 1474-175X.
- ^ Han, Eun-Soo; Muller, Florian L.; Pérez, Viviana I.; Qi, Wenbo; Liang, Huiyun; Xi, Liang; Fu, Chunxiao; Doyle, Erin; Hickey, Morgen (tháng 6 năm 2008). “The in vivo gene expression signature of oxidative stress”. Physiological Genomics (bằng tiếng Anh). 34 (1): 112–126. doi:10.1152/physiolgenomics.00239.2007. ISSN 1094-8341.
Wikimedia Commons có thêm hình ảnh và phương tiện truyền tải về P53.
Nguyễn ánh











































Chúng tôi bán

 GIẢM
0%
GIẢM
0%
438.000 ₫
438.000 ₫
 GIẢM
29%
GIẢM
29%
10.000 ₫
14.000 ₫
 GIẢM
13%
GIẢM
13%
6.500 ₫
7.500 ₫
 GIẢM
15%
GIẢM
15%
340.000 ₫
400.000 ₫
 GIẢM
-26%
GIẢM
-26%
140.000 ₫
111.000 ₫
Bài viết liên quan

Giới thiệu AG Adara - Magenta Meteor Artery Gear: Fusion
Sở hữu năng lực xoá buff diện rộng kèm hiệu ứng Speed Reduction, đặc biệt là rush action cực khủng

Sơn mài - hình thức nghệ thuật đắt giá của Việt Nam
Sơn mài là một hình thức tranh sơn phết truyền thống của Việt Nam được tạo ra từ một loại sơn độc được thu hoạch từ một vùng xa xôi của đất nước

Giới thiệu Light Novel: Isekai Meikyuu no Saishinbu wo Mezasou
Một chàng trai thành phố bất ngờ tỉnh lại trong một hành lang tối tăm mà không hiểu chuyện gì đang xảy ra.

Nền kinh tế tư nhân của Triều Tiên
Triều Tiên, một trong những nước có nền kinh tế “đóng” nhất trên thế giới, đang có những bước phát triển mạnh mẽ.